- CRYOLOGIE - Cryogénie
- CRYOLOGIE - CryogénieLa production de froid, ou extraction de chaleur, a pour but soit d’abaisser la température d’un milieu, et éventuellement, dans le cas de l’air, sa teneur en vapeur d’eau, soit de refroidir une substance au cours d’un processus de conservation ou de transformation.Pour la zone des températures de l’ambiance jusque vers 漣 100 0C (conditionnement de l’air, réfrigération et congélation des denrées, traitements industriels, etc.), la production de froid résulte, dans la très grande majorité des cas, d’une transition de phase (évaporation, condensation) d’un fluide pur ou d’un mélange, azéotropique ou non, de fluides convenablement choisis.1. Production du froidPour toute température comprise entre celle du point triple et celle du point critique d’un fluide pur, la pression de la vapeur en équilibre avec le liquide est la pression de vapeur saturante relevée sur la courbe des pressions de vapeur portée sur la figure 1. Pour qu’un tel fluide s’évapore dans un échangeur appelé évaporateur (fig. 1 et 2), il faut que sa température 0 (0C), ou T 0 (K), soit inférieure à la température i du milieuà refroidir – considéré comme la source froide – ; la chaleur latente d’évaporation est fournie par ce milieu, d’où: 0 麗 i . La pression régnant dans l’évaporateur est alors la pression p 0 de vapeur saturante, en l’absence de gaz inertes (rentrées d’air). La vapeur ainsi obtenue à basse pression et basse température peut être liquéfiée à la température c dans un second échangeur dénommé condenseur , refroidi par le milieu naturel extérieur (air ambiant, eau de ville, de puits, de mer, etc.), qui est la source chaude et dont la température ex est inférieure à c, à condition que la vapeur ait été comprimée au préalable jusqu’à la pression de condensation p c correspondant à c. Le liquide obtenu est ramené à l’évaporateur après avoir subi une détente, accompagnée d’un refroidissement depuis la pression p c jusqu’à p 0. Ce refroidissement consécutif à l’amortissement de l’accroissement d’énergie cinétique du fluide relève des lois de l’effet Joule-Thomson exposées à l’article THERMODYNAMIQUE (thermodynamique technique, conservation de l’enthalpie, conditions d’inversion). Cette opération s’accomplit sans travail utile extérieur et est assurée par un détendeur (robinet, orifice calibré, capillaire, etc.) et porte généralement le nom de détente par laminage.Un tel système constitue une machine frigorifique s’il est conçu en vue de mettre à profit la production de froid à la source froide, mais il est une pompe à chaleur lorsque le but recherché est l’utilisation de la chaleur libérée à la source chaude.Fluides frigorigènesLes fluides appelés fluides frigorigènes (symbole R) doivent répondre aux conditions fondamentales suivantes:– avoir des propriétés thermodynamiques satisfaisantes;– avoir de bonnes propriétés de transfert de chaleur et de masse;– être chimiquement stables et neutres à l’égard des constituants du circuit.De plus, il convient qu’un frigorigène soit sans toxicité ou aussi peu toxique que possible, qu’il soit non ou difficilement inflammable, qu’il ne pose pas de difficultés technologiques (pour la détection de fuite, pour la lubrification, à l’égard de la présence d’eau et d’air, etc.), enfin qu’il soit d’un prix acceptable. Jusqu’à un passé récent, l’industrie utilisait l’ammoniac, des chlorofluorocarbures totalement halogénés (C.F.C.) ou partiellement halogénés (H.C.F.C.) ainsi que l’eau; l’eau est notamment le frigorigène des machines à absorption pour la climatisation.Or, les études de la haute atmosphère ayant mis en évidence l’appauvrissement de la couche d’ozone strastosphérique par le chlore ainsi que par le brome, les autorités politiques ont adopté en 1987 le protocole de Montréal . Celui-ci limitait la production des composés néfastes pour l’ozone, parmi lesquels la quasi-totalité des frigorigènes halogénés (C.F.C.). Par la suite, des mesures plus sévères ont été prises à Londres en 1990, puis à Copenhague en 1992, prévoyant l’arrêt de la production des C.F.C. au plus tard en 1996; pour la Communauté européenne, la date limite est 1995. Pour les composés H.C.F.C., qui comportent de l’hydrogène dans leur molécule et sont de ce fait moins stables, la date limite de production est 2030. Compte tenu de ces décisions, l’industrie chimique a cherché des fluides de remplacement, ce qui nécessite un délai d’étude et de développement d’une quinzaine d’années. Ces nouvelles substances sont des halogénés non chlorés. Mais, vers les années 1985, les physiciens de l’atmosphère ont établi une relation entre l’augmentation de la teneur de l’atmosphère en « gaz industriels » (CO2, CH4NOx Oz troposphérique, C.F.C. et H.C.F.C.) et l’accroissement de la température de la planète. Il s’agit de l’effet de serre . En particulier, les frigorigènes de remplacement, qui sont des fluorocarbures, contribuent de façon significative à cet effet: une molécule de ces composés est équivalente, selon sa composition, à plusieurs milliers de molécules de C2.On peut penser que, dans le futur, des mesures restrictives de production de tout composé halogéné seront adoptées; s’il en était ainsi, les systèmes frigorifiques actuellement en usage devraient être très profondément modifiés. Deux axes d’évolution sont prévisibles:– recherche de frigorigènes sans effet sur l’ozone et sans effet de serre pour le système à compression mécanique;– mise au point et développement d’autres systèmes frigoriques (à sorption, à air, à machine Stirling, etc.).L’ammoniac (ou R717) a été un des premiers fluides utilisés. Il a de bonnes propriétés thermodynamiques et de transfert de chaleur. Toutefois, sa température au refoulement du compresseur peut être excessive pour les huiles frigorifiques qui s’altèrent à partir de 130 0C. Aussi, il est nécessaire de prendre certaines précautions, si la surchauffe au refoulement est trop grande (cf. infra , Système bi-étagé ).L’ammoniac est un fluide peu coûteux, mais son emploi n’est pas sans danger : il peut former avec l’air un mélange explosif; il provoque des brûlures aux yeux et à la peau si elle est humide; l’air pollué par l’ammoniac est suffocant; il s’est déjà produit un certain nombre d’accidents mortels dus à des fuites importantes de ce fluide. Aussi, l’ammoniac est essentiellement employé dans les équipements industriels de traitement et de conservation de denrées périssables (produits réfrigérés, congelés et surgelés).Cependant, l’ammoniac étant sans effet néfaste sur l’ozone et ne participant pas au réchauffement de la planète, la construction frigorifique développe des « petits » compresseurs, notamment pour les besoins des commerces.Les frigorigènes halogénés réglementés par le protocole de Montréal sont des chlorofluorocarbures , dérivés principalement du méthane et de l’éthane, dont les atomes d’hydrogène sont, totalement ou non, remplacés par des atomes de chlore et de fluor. Leur dénomination générale est celle de R (de l’anglais refrigerant ) suivi d’un chiffre caractéristique de leur composition. Ils sont utilisés à l’état pur ou en mélange azéotropique, ou encore pseudo-azéotropique. Ils assurent tous les niveaux de température des machines frigorifiques (y compris des réfrigérateurs ménagers) et des pompes à chaleur. À la pression atmosphérique, la température d’évaporation va de 漣 128 0C, pour le R14, à + 47,6 0C, pour le R113, et est, pour les fluides les plus courants, de 漣 30 0C pour le R12, de 漣 41 0C pour le R22 et de 漣 45,6 0C pour le mélange azéotropique R502.Les halogénés, entièrement halogénés sans odeur, non toxiques et ne brûlant pas, étaient employés dans les équipements fonctionnant dans des locaux fréquentés par le public (bureaux, salles de spectacles, locaux d’habitation et commerciaux). Les machines frigorifiques de la chaîne du froid, mis à part les gros entrepôts, fonctionnaient en très grande majorité avec des systèmes à compression de chlorofluorocarbures.Les fluides de remplacement non réglementés sont sans chlore et sont partiellement hydrogénés. Ainsi, le substitut du R12 – le fluide le plus utilisé, et dont la formule est CCl22 – est le R134a de formule C2H24. Il est inoffensif pour l’ozone; par contre, son effet de serre serait équivalent à 1 200 molécules de C2.Enfin, l’emploi de ce fluide a soulevé quelques difficultés technologiques concernant la lubrification des compresseurs, le choix des vernis des moteurs électriques pour les machines hermétiques et la détection des fuites. De plus, le coefficient de performance (COP) des machines à R134a est inférieur à celui des machines à R12 de l’ordre de 15 à 20 p. 100 aux températures de la congélation. L’arrêt de production des fluides réglementés pose le problème de l’entretien et de la réparation des machines en service, surtout si elles ne sont pas financièrement amorties. Le moyen mis en œuvre, et qui est rendu obligatoire, est la récupération et le recyclage des fluides usagés, ce qui permet de prolonger la période d’utilisation des équipements et ce qui réduit les rejets dans l’atmosphère.Diagrammes thermodynamiquesSi la relation (p , ), qui est caractéristique d’un fluide, indique quelles sont les pressions et températures de fonctionnement d’un système frigorifique, elle ne fournit aucun renseignement sur les quantités d’énergie mises en jeu (fig. 1).Système mono-étagé. Le diagramme enthalpique ou de Mollier (lg p ,i ), où i est l’enthalpie massique (notée aussi h en thermodynamique) et p la pression de vapeur saturante, établi pour 1 kilogramme d’un fluide défini, pallie cette lacune et permet de dessiner le cycle d’un système frigorifique à compression (fig. 3).Pour assurer la puissance frigorifique W 0, dans des conditions données de fonctionnement, il faut que le débit massique d 0 corresponde à la valeur:
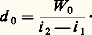
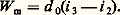 La puissance rejetée au condenseur W c est:
La puissance rejetée au condenseur W c est: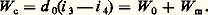 Les relations ci-dessus sont les expressions permettant de calculer les éléments d’un système frigorifique.Système bi-étagé. La production d’une machine à un étage de compression diminue notablement aux basses températures d’évaporation (congélation par exemple). De plus, avec l’ammoniac, la température de refoulement dépasse 130 0C dès que l’écart entre p c et p 0 croît, ce qui dégrade l’huile car celle-ci est de qualité spéciale.Pour pallier ces difficultés, on adopte une machine à deux étages: un premier compresseur comprime les vapeurs de p 0 à une pression intermédiaire p m; et un second de p m à p c, après désurchauffe des vapeurs. Il existe plusieurs systèmes à deux étages; le système à injection totale et à évaporateur alimenté par regorgement et gravité est l’un de ceux utilisés pour les équipements industriels de congélation et, de façon plus générale, les équipements à basse température.RendementsLe rendement 兀 d’une machine frigorifique, ou d’une pompe à chaleur, exprime le rapport de la production de froid (évaporateur) ou de chaleur (condenseur) à la dépense en énergie de compression nécessaire à ces productions.Pour une machine parfaite fonctionnant entre les températures absolues T sc de source chaude et T sf de source froide, le rendement maximal ou de Carnot 兀 max vaut:
Les relations ci-dessus sont les expressions permettant de calculer les éléments d’un système frigorifique.Système bi-étagé. La production d’une machine à un étage de compression diminue notablement aux basses températures d’évaporation (congélation par exemple). De plus, avec l’ammoniac, la température de refoulement dépasse 130 0C dès que l’écart entre p c et p 0 croît, ce qui dégrade l’huile car celle-ci est de qualité spéciale.Pour pallier ces difficultés, on adopte une machine à deux étages: un premier compresseur comprime les vapeurs de p 0 à une pression intermédiaire p m; et un second de p m à p c, après désurchauffe des vapeurs. Il existe plusieurs systèmes à deux étages; le système à injection totale et à évaporateur alimenté par regorgement et gravité est l’un de ceux utilisés pour les équipements industriels de congélation et, de façon plus générale, les équipements à basse température.RendementsLe rendement 兀 d’une machine frigorifique, ou d’une pompe à chaleur, exprime le rapport de la production de froid (évaporateur) ou de chaleur (condenseur) à la dépense en énergie de compression nécessaire à ces productions.Pour une machine parfaite fonctionnant entre les températures absolues T sc de source chaude et T sf de source froide, le rendement maximal ou de Carnot 兀 max vaut: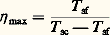 pour une machine frigorifique, et:
pour une machine frigorifique, et: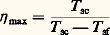 pour une pompe à chaleur.En fait, 0 est inférieure de 0 à la température de la source froide, et c supérieure de c à celle de la source chaude. 0 et c étant définies, on peut tracer dans le cas d’une machine réelle mono-étagée et pour un fluide donné le cycle théorique (fig. 3) et déterminer son rendement théorique 兀 th, ce qui donne:
pour une pompe à chaleur.En fait, 0 est inférieure de 0 à la température de la source froide, et c supérieure de c à celle de la source chaude. 0 et c étant définies, on peut tracer dans le cas d’une machine réelle mono-étagée et pour un fluide donné le cycle théorique (fig. 3) et déterminer son rendement théorique 兀 th, ce qui donne: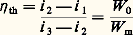 pour une machine frigorifique, et:
pour une machine frigorifique, et: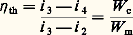 pour une pompe à chaleur.Dans un système à compression, le travail pour élever la pression de p 0 à p c est fourni par un ensemble moteur-transmission-compresseur, dont le rendement global est 兀 g. Le rendement, ou coefficient de performance COP du système, est:
pour une pompe à chaleur.Dans un système à compression, le travail pour élever la pression de p 0 à p c est fourni par un ensemble moteur-transmission-compresseur, dont le rendement global est 兀 g. Le rendement, ou coefficient de performance COP du système, est: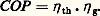 Enfin, on peut exprimer le COP par rapport à l’énergie primaire. Si le rendement de la transformation de l’énergie primaire en énergie fournie au moteur du compresseur est 兀 E, le rendement COP E est:
Enfin, on peut exprimer le COP par rapport à l’énergie primaire. Si le rendement de la transformation de l’énergie primaire en énergie fournie au moteur du compresseur est 兀 E, le rendement COP E est: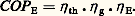 Le rendement énergétique des systèmes frigorifiques est d’autant meilleur que c 漣 0 est plus faible.Machines frigorifiquesLa compression est assurée par un motocompresseur ou un dispositif à sorption.Systèmes à compressionLes compresseurs sont soit volumétriques (polycylindriques à pistons, rotatifs, à vis ou à spirales), soit du type turbomachine. Les premiers peuvent fournir un taux important de compression. Les turbocompresseurs, le plus souvent centrifuges, comportent une roue à aubages qui, en tournant à une vitesse suffisante (de 3 000 à 30 000 tr/min), élève la pression du fluide; pour augmenter le taux de compression, on associe plusieurs roues en série.La puissance frigorifique (ou calorifique) d’un équipement est, dans des conditions définies de pression, proportionnelle au débit massique du fluide; les compresseurs peuvent être classés en fonction des puissances frigorifiques (ou calorifiques) des circuits qu’ils équipent: de 0,1 à 500 kilowatts pour les compresseurs polycylindriques rotatifs ou à spirales, de 0,5 à 1,5 mégawatt pour ceux à vis et de plus de 1 mégawatt pour les turbocompresseurs. Le groupe motocompresseur, constitué du compresseur et du moteur qui l’entraîne, comprend en général un moteur électrique, mais parfois ce dernier est thermique dans certains équipements à énergie totale ou dans les machines mobiles (engins frigorifiques de transport).Un groupe motocompresseur, à moteur électrique, est ouvert si les deux machines sont constamment accessibles. Pour les fluides halogénés, qui sont très sensibles à l’humidité et dont la tension superficielle est faible, l’ensemble motocompresseur peut être contenu dans un carter étanche. Si celui-ci est soudé, aucune réparation n’est possible (cas des réfrigérateurs ménagers): le groupe est hermétique . S’il est démontable, moteur et compresseur sont réparables: le groupe est appelé semi-hermétique.Pour les systèmes à deux étages, un seul compresseur polycylindrique à pistons, dit compound , peut assurer les fonctions du compresseur basse pression et du compresseur haute pression. Si le nombre de cylindres est un multiple de 3, les deux tiers, et s’il est un multiple de 4, les trois quarts des cylindres servent à la compression de p 0 à la moyenne pression p m.Système à sorptionLes systèmes à sorption sont fondés sur la différence de fixation du frigorigène dans un liquide (absorption) ou sur un solide (adsorption). Dans le cas de l’adsorption, le phénomène peut être purement physique comme cela est le cas de [eau-zéolite] ou [méthanol-charbon actif]; il peut résulter d’une réaction chimique réversible: comme les ammoniacates (ammoniac-chlorure) ou les hydrures (hydrogène-métal).Le procédé actuellement le plus utilisé est celui à absorption, qui est décrit ci-dessous. Cependant, dans l’avenir, il est vraisemblable que certains équipements à adsorption seront développés industriellement.Le système à absorption , aux températures inférieures à 0 0C, utilise le couple [ammoniac-eau]: les vapeurs d’ammoniac, à p 0, sortant de l’évaporateur sont absorbées par de l’eau contenue dans un récipient, l’absorbeur, qui est refroidi, car le phénomène est exothermique. La solution riche ainsi formée est pompée dans un autre récipient, le bouilleur, ou générateur, qui est chauffé, de sorte que l’ammoniac, étant moins soluble à chaud, se sépare à l’état de vapeur à la pression p c et passe dans le condenseur. La solution pauvre résiduelle revient à l’absorbeur après détente et échange de chaleur avec la solution riche (fig. 4).On peut représenter l’évolution de la solution au moyen d’un diagramme (lg p ,1/T ) [T température absolue], où les isotitres du liquide et de la vapeur forment deux familles de droites.Le cycle 1, 2, 3, 4 (fig. 5) montre que le système est d’autant plus efficace que B et p 0 sont plus grands et p c plus petit. Il existe une limite Bmin, pour B, quand les isotitres x R et x p sont confondus. Quand x R tend vers x p, il faut adopter un système à deux étages.Le rendement maximal (ou de Carnot) 兀 max est le produit du rendement d’un moteur thermique parfait fonctionnant entre T B et T c (correspondant à B et c) et de celui d’une machine frigorifique parfaite, entre T c et T 0 (correspondant à 0):
Le rendement énergétique des systèmes frigorifiques est d’autant meilleur que c 漣 0 est plus faible.Machines frigorifiquesLa compression est assurée par un motocompresseur ou un dispositif à sorption.Systèmes à compressionLes compresseurs sont soit volumétriques (polycylindriques à pistons, rotatifs, à vis ou à spirales), soit du type turbomachine. Les premiers peuvent fournir un taux important de compression. Les turbocompresseurs, le plus souvent centrifuges, comportent une roue à aubages qui, en tournant à une vitesse suffisante (de 3 000 à 30 000 tr/min), élève la pression du fluide; pour augmenter le taux de compression, on associe plusieurs roues en série.La puissance frigorifique (ou calorifique) d’un équipement est, dans des conditions définies de pression, proportionnelle au débit massique du fluide; les compresseurs peuvent être classés en fonction des puissances frigorifiques (ou calorifiques) des circuits qu’ils équipent: de 0,1 à 500 kilowatts pour les compresseurs polycylindriques rotatifs ou à spirales, de 0,5 à 1,5 mégawatt pour ceux à vis et de plus de 1 mégawatt pour les turbocompresseurs. Le groupe motocompresseur, constitué du compresseur et du moteur qui l’entraîne, comprend en général un moteur électrique, mais parfois ce dernier est thermique dans certains équipements à énergie totale ou dans les machines mobiles (engins frigorifiques de transport).Un groupe motocompresseur, à moteur électrique, est ouvert si les deux machines sont constamment accessibles. Pour les fluides halogénés, qui sont très sensibles à l’humidité et dont la tension superficielle est faible, l’ensemble motocompresseur peut être contenu dans un carter étanche. Si celui-ci est soudé, aucune réparation n’est possible (cas des réfrigérateurs ménagers): le groupe est hermétique . S’il est démontable, moteur et compresseur sont réparables: le groupe est appelé semi-hermétique.Pour les systèmes à deux étages, un seul compresseur polycylindrique à pistons, dit compound , peut assurer les fonctions du compresseur basse pression et du compresseur haute pression. Si le nombre de cylindres est un multiple de 3, les deux tiers, et s’il est un multiple de 4, les trois quarts des cylindres servent à la compression de p 0 à la moyenne pression p m.Système à sorptionLes systèmes à sorption sont fondés sur la différence de fixation du frigorigène dans un liquide (absorption) ou sur un solide (adsorption). Dans le cas de l’adsorption, le phénomène peut être purement physique comme cela est le cas de [eau-zéolite] ou [méthanol-charbon actif]; il peut résulter d’une réaction chimique réversible: comme les ammoniacates (ammoniac-chlorure) ou les hydrures (hydrogène-métal).Le procédé actuellement le plus utilisé est celui à absorption, qui est décrit ci-dessous. Cependant, dans l’avenir, il est vraisemblable que certains équipements à adsorption seront développés industriellement.Le système à absorption , aux températures inférieures à 0 0C, utilise le couple [ammoniac-eau]: les vapeurs d’ammoniac, à p 0, sortant de l’évaporateur sont absorbées par de l’eau contenue dans un récipient, l’absorbeur, qui est refroidi, car le phénomène est exothermique. La solution riche ainsi formée est pompée dans un autre récipient, le bouilleur, ou générateur, qui est chauffé, de sorte que l’ammoniac, étant moins soluble à chaud, se sépare à l’état de vapeur à la pression p c et passe dans le condenseur. La solution pauvre résiduelle revient à l’absorbeur après détente et échange de chaleur avec la solution riche (fig. 4).On peut représenter l’évolution de la solution au moyen d’un diagramme (lg p ,1/T ) [T température absolue], où les isotitres du liquide et de la vapeur forment deux familles de droites.Le cycle 1, 2, 3, 4 (fig. 5) montre que le système est d’autant plus efficace que B et p 0 sont plus grands et p c plus petit. Il existe une limite Bmin, pour B, quand les isotitres x R et x p sont confondus. Quand x R tend vers x p, il faut adopter un système à deux étages.Le rendement maximal (ou de Carnot) 兀 max est le produit du rendement d’un moteur thermique parfait fonctionnant entre T B et T c (correspondant à B et c) et de celui d’une machine frigorifique parfaite, entre T c et T 0 (correspondant à 0):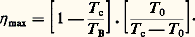 Théoriquement, un système à absorption est aussi efficace qu’un système à compression avec son moteur, pourvu que les trois températures de source se correspondent respectivement.Pour les températures supérieures à 0 0C (climatisation), on peut employer le couple eau-bromure de lithium, dans lequel l’eau est le fluide frigorigène.Si on introduit dans l’évaporateur une pression partielle p d’hydrogène de sorte que p c = p 0 + p , on élimine la pompe de circulation, et la solution circule par thermosiphon: c’est le cas des réfrigérateurs ménagers.Les systèmes à absorption sont généralement adoptés lorsqu’on ne dispose pas d’énergie mécanique, ou pour valoriser de la chaleur perdue (vapeur détendue industrielle).Évaporation et condensationÉvaporateursLes évaporateurs sont des refroidisseurs d’air ou de liquide et sont alimentés par injection directe ou regorgement.Les refroidisseurs d’air sont appelés frigorifères. L’évaporateur, compris dans un caisson, est constitué de tubes à ailettes dont l’écartement est d’autant plus grand que 0 est plus basse: de 8 à 12 millimètres en réfrigération et de 16 à 20 millimètres en congélation. L’air, aspiré par des ventilateurs en bas du local, est refroidi sur l’évaporateur et est refoulé sous le plafond (l’évaporateur des réfrigérateurs ménagers fonctionne en convection naturelle et est constitué de plaques d’aluminium).Les refroidisseurs de liquide (pour les industries alimentaires, ou pour les circuits de climatisation à eau glacée) sont constitués d’un évaporateur à tubes lisses placé dans un bac ou dans un échangeur, de conception assez semblable à celle d’un condenseur multitubulaire.Alimentation des évaporateursEn injection directe , le fluide, détendu de p c à p 0, est directement introduit dans l’évaporateur; son débit est réglé par le détendeur (détendeur thermostatique) pour qu’en sortant il soit surchauffé. S’il n’en était pas ainsi, le compresseur pourrait aspirer du liquide, et les clapets d’aspiration risqueraient de casser (coup de liquide ); en outre, s’il y a entraînement d’huile, la vitesse de la vapeur basse pression doit être d’au moins 10 mètres par seconde, pour ramener l’huile au compresseur.Ce système est simple et utilisable avec tout fluide, pour des machines de toutes puissances; cependant, en pratique, il est surtout réservé aux équipements de petite et moyenne puissance (appareils commerciaux, agricoles, petits climatiseurs). En effet, l’efficacité de l’évaporateur ne peut pas être maximale, car de la vapeur apparaît dès la détente, or les transferts sont moins bons en phase gazeuse qu’en phase liquide.Dans l’alimentation par regorgement , le fluide est détendu dans une bouteille séparatrice basse pression (B.S.B.P.); la vapeur, qui est saturée sèche, est aspirée en haut de cette bouteille, tandis que le liquide basse pression s’accumule dans le fond et est envoyé, par gravité ou pompe, dans l’évaporateur. Le fluide, sortant de ce dernier, a un excès de liquide; l’efficacité de l’évaporateur est d’autant meilleure que le titre en liquide du fluide au retour dans la B.S.B.P. est plus grand, ce qui explique l’intérêt de l’alimentation par pompe. Le détendeur, à flotteur ou électronique, règle le niveau liquide dans la B.S.B.P.L’alimentation par regorgement est réservée aux équipements industriels de grosse puissance et à ceux à régime thermique variable (tunnels de congélation, par exemple).CondenseursSelon le milieu utilisé, le condenseur est à air ou à eau. Les condenseurs à air des réfrigérateurs ménagers sont à convection naturelle; mais tous les autres appareils, de puissance commerciale ou industrielle, sont à convection forcée: l’air est aspiré par des ventilateurs à travers une batterie à ailettes serrées; le débit est important: exprimé en mètres cubes par heure, le chiffre est à peu près quatre à cinq fois plus grand que celui de la puissance frigorifique en watts.Le condenseur à eau en circuit ouvert le plus utilisé est du type multitubulaire horizontal, où l’eau fait un ou plusieurs passages dans des tubes, tandis que le fluide se condense dans l’espace compris entre l’extérieur des tubes et la virole. Comme l’échauffement de l’eau est de l’ordre de 5 à 10 0C, le débit d’eau en litres par heure est voisin du chiffre de la puissance en watts divisé par . Pour réduire la consommation, l’eau est recyclée dans un réfrigérant atmosphérique , où elle est refroidie par évaporation. La dépense est, en théorie, égale à 1 ou 2 p. 100 et, en pratique, à 5 p. 100 du débit en circuit ouvert car, pour limiter la concentration en sels, une partie de l’eau est purgée. Le condenseur et le réfrigérant atmosphérique peuvent être réunis en un seul appareil, qui est un condenseur évaporatif. Notons que, en France, les installations à condenseur à eau doivent comporter un dispositif économiseur.ApplicationsActuellement, pratiquement dans toutes les activités humaines, industrielles, agricoles, sociales, médicales, etc., interviennent des systèmes frigorifiques à fluide liquéfiable.L’application la plus ancienne concerne la conservation des denrées périssables. En 1876, le Français Charles Tellier démontrait de façon spectaculaire les possibilités du froid artificiel en transportant des carcasses de viande fraîche de Rouen à Buenos Aires.De nos jours, les techniques frigorifiques sont applicables pratiquement à toutes les denrées périssables, aux produits manufacturés altérables et tout spécialement aux plats élaborés ou cuisinés. À ce propos, il convient de souligner le succès dans tous les pays industrialisés des produits congelés et surtout surgelés. Cette dernière qualification est réservée à des produits d’excellente qualité initiale, sains, congelés rapidement jusqu’à une température de 漣 18 0C à cœur, emballés, conservés, transportés et commercialisés – en permanence à 漣 18 0C ou moins –, jusqu’à leur livraison en l’état aux consommateurs.Le respect de ces spécifications à l’échelle industrielle, clairement définies par le décret du 9 septembre 1964, est l’une des raisons du succès des produits surgelés dont la consommation en France progresse de 10 à 20 p. 100 par an depuis une vingtaine d’années. Les autres raisons majeures sont sans doute, d’une part, le prix de revient de la congélation industrielle (c’est-à-dire préalable à la distribution), qui est du même ordre de grandeur que celui de l’apertisation, et, d’autre part, les besoins énergétiques, qui sont à peu près deux fois plus petits que pour l’apertisation, et, enfin, la facilité d’emploi et la rapidité de préparation. Il est certain que la France n’a pu devenir fortement exportatrice pour toute une série de productions périssables que grâce au froid. Aussi, son équipement, réalisé depuis la Libération, est l’un des plus importants et des plus modernes du monde.Parallèlement au froid alimentaire, les techniques du conditionnement d’air se sont développées et diversifiées pour le confort de l’homme (bureaux, hôpitaux, salles de spectacles, logements, etc.) et pour la maîtrise de l’ambiance de certains locaux (d’ordinateur par exemple), par le contrôle de la température et de l’humidité relative, le filtrage d’air dans les industries textiles, papetières, de mécanique de précision, etc. Le froid est également un facteur de sécurité. Ainsi, toute centrale nucléaire comprend, pour cette raison, des machines frigorifiques puissantes.Une autre illustration remarquable des techniques frigorifiques concerne les progrès considérables accomplis en médecine et en chirurgie. Les greffes d’organes, et notamment les greffes cardiaques, impliquent son intervention. La préparation du plasma lyophilisé, qui a sauvé depuis la Seconde Guerre mondiale un grand nombre de vies humaines, comporte une phase initiale de congélation. Actuellement se développe une nouvelle chirurgie, la cryochirurgie, qui, par la congélation des tissus malades avec une sonde à basse température, permet leur ablation tout en ménageant les tissus sains avoisinant. Elle a eu de remarquables succès pour la guérison de certains cancers et tumeurs (peau, cerveau, poumons, etc.).La cryogénie a permis l’essor du trafic maritime des gaz naturels et gaz de pétrole, grâce à leur stockage à l’état liquide à la pression atmosphérique (transport par « navires méthaniers »). La cryogénie fournit également le carburant liquide nécessaire au lancement des fusées. En outre, pour l’avenir, les perspectives d’industrialisation de l’effet supraconducteur à « haute température », découvert en 1987, révèlent des possibilités d’application d’une portée naguère insoupçonnée.Enfin, par suite de la crise pétrolière, la recherche de procédés plus économes en énergie ou de récupération de chaleur, ainsi que l’exploitation de l’énergie solaire ont mis en évidence l’intérêt des pompes à chaleur, des transformateurs de chaleur et des systèmes à recompression mécanique des vapeurs qui trouvent leur origine dans la technique frigorifique. Grâce à ces procédés, le rendement de raffinage du brut pétrolier est passé de 94 à 98 p. 100.En conclusion, le froid artificiel, bien qu’il soit déjà plus que centenaire, fait encore partie aujourd’hui des techniques de pointe, en perpétuelle évolution.2. Liquéfaction et séparation des gazOn liquéfie des gaz par refroidissement à basse température soit pour obtenir des liquides directement utilisables en tant que fluide cryogénique ou parce que leur compacité facilite le stockage ou le transport, soit parce que la production du liquide constitue une étape intermédiaire permettant la séparation des composants du gaz liquéfié par condensation fractionnée ou par distillation.La liquéfaction des gaz est entrée dans sa phase industrielle au début de ce siècle grâce aux travaux de Karl von Linde et de Georges Claude. Son développement, d’abord assez lent, n’a fait que s’accélérer depuis, et une industrie très puissante et très dynamique, ayant ses techniques propres, repose sur les bases posées par ces chercheurs. On peut s’étonner que l’obtention aisée des basses températures, entre 漣 40 0C et 漣 210 0C, soit à l’origine d’une activité industrielle aussi importante. Il faut en trouver la raison dans le fait que les fameux « gaz permanents » du XIXe siècle, qui constituent les matières premières essentielles de cette industrie, sont des éléments fondamentaux puisqu’il s’agit principalement des gaz de l’air, de l’hydrogène, de l’oxyde de carbone, du méthane et du bioxyde d’azote.La séparation des mélanges gazeux peut être réalisée par différentes techniques: l’adsorption (en particulier l’adsorption à température ambiante et pression variable, dite Pressure Swing Adsorption ou P.S.A., qui s’est beaucoup développée ces dernières années), la perméation et la distillation.En fait, la technique de distillation, qui s’est développée la première, tient encore aujourd’hui une place prépondérante.Les progrès technologiques réalisés au cours des vingt dernières années ont permis de ramener les pertes thermiques, conséquence d’une distillation à basse température, à des valeurs très faibles. Ainsi, pour un appareil de séparation d’air de grande taille, la tenue frigorifique ne requiert que 6 à 7 p. 100 de l’énergie totale dépensée pour la séparation elle-même. Dans ce cas, les échangeurs de chaleur présentent des écarts minimaux de température de 1 à 3 0C, et les plateaux de distillation, des pertes de charge inférieures à 2,5 cm d’eau. Depuis 1985, une nouvelle technique de distillation par garnissage organisée s’est progressivement développée; elle permet de réduire la perte de charge par plateau théorique à moins de 1 cm d’eau.L’emploi du chalumeau oxyacétylénique, mis au point par Charles Picard en 1901, est à l’origine d’une production industrielle de l’oxygène, encore bien modeste à l’époque. La capacité des appareils de production augmenta progressivement entre les deux guerres mondiales pour atteindre quelques centaines à quelques milliers de mètres cubes par heure.L’introduction, puis la généralisation, des procédés d’élaboration de l’acier à l’oxygène a pratiquement décuplé la production d’oxygène au cours des années 1960-1970.De nouvelles utilisations de l’oxygène dans l’industrie chimique (oxyde d’éthylène, cracking d’hydrocarbures, gazéification du charbon) ont conduit à augmenter sans cesse la taille des installations.Les plus grandes unités actuelles ont été réalisées par L’Air liquide pour la gazéification du charbon dans les usines de Sasol en Afrique du Sud. La centrale d’oxygène comprend treize lignes de production de 2 250 t/j d’oxygène à une pureté de 98,5 p. 100 et à une pression de 3,5 MPa.L’important développement de réseaux de distribution par gazoduc dans les pays industrialisés, la sécurité d’approvisionnement assurée par des stockages liquides de grande capacité (de 3 000 000 à 4 000 000 l) font de l’oxygène une utilité fiable, et d’une grande souplesse d’utilisation.Parallèlement au développement industriel de l’oxygène extrait de l’air atmosphérique, l’essor simultané de l’azote obtenu dans les mêmes installations n’a pas été moins impressionnant.Les premières applications ont été la production d’ammoniac par synthèse sur catalyseur à haute pression. L’ammoniac, comme l’on sait, est constitué de trois volumes d’hydrogène pour un volume d’azote, et, dès les premières installations réalisées après la mise au point du procédé de synthèse par Haber en 1913, l’azote fut produit par distillation d’air à basse température, et l’hydrogène provenant d’un gaz à l’eau, ou d’un gaz de fours à coke, fut lui aussi purifié par refroidissement à basse température, puis « lavage à l’azote liquide ».À partir de la production d’ammoniac et de celle d’acide nitrique (R 漣H3) qui résulte de son oxydation, une vaste industrie des engrais azotés a pris naissance. Son développement rapide devait bientôt libérer l’Europe de son assujettissement aux engrais naturels étrangers, par exemple les nitrates du Chili. L’acide nitrique concentré trouvait également des débouchés dans le domaine militaire (explosifs, vésicants).Ainsi, historiquement, la liquéfaction de l’air apparaît comme l’une des contributions fondamentale à l’essor de l’industrie chimique lourde contemporaine [cf. GÉNIE CHIMIQUE].Mais l’azote, en temps que gaz neutre à bon marché, disponible si nécessaire en très grandes quantités, trouve aujourd’hui une multitude d’applications, et sa consommation ne cesse de croître.De même, l’argon, gaz rare non réactif présent dans l’atmosphère, produit simultanément à l’oxygène, voit son marché s’accroître constamment, en particulier dans le domaine de la soudure et de la sidérurgie.Outre le traitement de l’air et la purification de gaz destinés à la synthèse de l’ammoniac, les séparations cryogéniques sont nombreuses. On les rencontre par exemple:– pour purifier l’hydrogène présent dans les gaz de raffineries et dont celles-ci ont besoin pour les procédés d’hydrogénation (en particulier pour la désulfuration);– pour purifier l’hydrogène et l’oxyde de carbone dans l’industrie chimique (synthèse de l’acide acétique);– pour produire les énormes quantités d’éthylène pur nécessaires à la fabrication des matières plastiques.À côté de la séparation des gaz, leur liquéfaction joue aussi un rôle de premier plan. La diminution de volume consécutive à la liquéfaction permet en effet de stocker et de transporter facilement, à la pression atmosphérique, l’oxygène, l’azote, l’argon et l’hélium, ainsi que l’hydrogène, le méthane et l’éthylène. L’emploi de l’hydrogène liquide comme combustible des fusées spatiales a conduit les États-Unis à construire d’énormes installations de production d’hydrogène liquide. Le transport de l’hélium liquide des États-Unis vers l’Europe est devenu d’une pratique courante, malgré la température d’ébullition très basse de l’hélium (face=F0019 漣 268,9 0C), et les techniques d’isolation particulièrement coûteuses pour réduire les pertes durant le transport.L’exemple le plus spectaculaire est cependant celui de la liquéfaction du gaz naturel. Parmi les sources d’énergie, le gaz naturel est le plus difficile à exploiter par suite de l’éloignement entre pays producteurs et pays consommateurs.Lorsqu’une liaison par gazoduc est impossible du fait d’une traversée maritime, on peut envisager le transport du gaz sous forme liquéfiée à la pression atmosphérique à 漣 161 0C dans des « méthaniers ».Les usines modernes ont plusieurs lignes de liquéfaction, chacune capable de liquéfier 400 000 nm3/h de gaz. Les méthaniers ont une capacité de 135 000 m3 en eau. Les tanks fixes de stockage au départ et à l’arrivée ont une capacité du même ordre.Le trafic entre l’Alaska (Shell), Brunei, l’Indonésie, île de D s et le Japon est devenu important, de même qu’entre l’Algérie et l’Europe.Mais il existe aussi une cinquantaine d’installations d’écrêtement des pointes de consommation (peak shaving en anglais). Les gazoducs de gaz naturel, peu chargés en été, sont incapables d’assurer les pointes de consommation hivernales, dont la durée est souvent limitée à quelques jours, voire à quelques semaines. On y remédie en liquéfiant et en stockant le gaz naturel au voisinage immédiat des lieux de consommation pendant la période estivale; ensuite, on revaporise le gaz naturel ainsi stocké pendant les pointes de consommation hivernales.Production des températures de liquéfactionCycle à cascadeLe moyen de production de froid par cascade repose sur le principe du cycle frigorifique à compression de vapeur et changement de phases (cf. CRYOLOGIE - Cryogénie [production du froid]).Le cycle à cascade consiste essentiellement en une suite de cycles de réfrigération utilisant chacun le fluide frigorigène le mieux adapté afin de transférer les frigories dont on a besoin de niveau en niveau jusqu’à la température finale désirée (fig. 6).Cycle à cascade incorporéeDans le cas du cycle à cascade incorporée, les différents compresseurs du cycle à cascade sont remplacés par une machine unique qui comprime un fluide frigorigène constitué par un mélange de gaz (fig. 7). La comparaison entre le schéma de principe d’un cycle à cascade classique et celui d’un cycle à cascade incorporée met en évidence la supériorité de ce dernier, non seulement du point de vue de la simplicité, de la sécurité d’exploitation et de l’entretien mais aussi du point de vue économique.Les premières applications de ce procédé, à l’échelle industrielle, ont été réalisées à Marsa el-Brega (Libye) dès 1970 par l’Air Product, et à Skikda (Algérie) par Technip-Air liquide, où sont traités avec succès 4,5 milliards de mètres cubes de gaz naturel par an.Ce procédé est aussi dénommé procédé à « fluide frigorigène composite » ou parfois à « fluide unique ». Il a reçu de nombreuses améliorations, notamment grâce à l’apport d’une réfrigération en tête au moyen d’un cycle à propane. Il est maintenant appliqué dans la quasi-totalité des installations de liquéfaction de gaz naturel.DétenteDeux procédés de détente existent: la détente sans travail utile extérieur évoquée dans la section précédente (cf. THERMODYNAMIQUE - Thermodynamique technique), essentiellement utilisée dès l’origine dans le procédé de Karl von Linde, et la détente avec fourniture de travail par l’intermédiaire d’une machine à pistons, selon le procédé de Georges Claude. Ce procédé, plus difficile à réaliser par suite de la présence d’organes mobiles aux basses températures de l’air liquide, mais susceptible d’un meilleur rendement énergétique, s’est imposé progressivement à la suite des perfectionnements successifs de la technique (lubrification à basse température, superfinition et pistons secs, mise au point des turbines, etc.).Dans les deux cas, on favorise la liquéfaction en faisant précéder le dispositif de détente par un échangeur de chaleur à contre-courant dans lequel le gaz détendu à basse température refroidit méthodiquement le gaz chaud à haute pression.Le cycle de StirlingUn procédé de production de froid par détente avec travail extérieur fonctionnant en circuit fermé est le cycle de Stirling. Dans ce cycle, le piston du compresseur et le piston de la machine de détente sont calés à 900 sur le même arbre-manivelle, la capacité de compression et la capacité de détente étant constamment en liaison par l’intermédiaire d’un régénérateur. L’ensemble se présente donc sous une forme très compacte, sans soupape de distribution. La société Philips a développé plusieurs modèles de machines frigorifiques utilisant ce cycle d’un rendement avantageux.Séparation des constituants de l’airL’air est un mélange de corps purs constitué essentiellement des gaz indiqués dans le tableau ci-dessous.Les constituants essentiels sont visiblement l’oxygène et l’azote. Viennent ensuite quelques gaz rares à teneur constante, puis un certain nombre d’impuretés à teneur variable et dépendant en particulier de la pollution: vapeur d’eau, C2 (moyenne 300 vpM), CH4 (moyenne 5 vpM), H2 etc.DistillationLa séparation de l’air en ses constituants s’effectue par distillation. Après compression et purification, l’air traité est refroidi jusqu’à sa température de rosée, pour y être distillé (fig. 8).Les courbes représentatives sur les figures 9 et 10 donnent, pour une pression donnée:– la correspondance entre la teneur en oxygène de la phase liquide et la teneur en oxygène de la phase vapeur en équilibre;– l’évolution de la teneur en oxygène de plateau théorique en plateau théorique, dans la colonne à moyenne pression (0,6 MPa abs.) et dans la colonne à basse pression (0,14 MPa abs.).Pour se rapprocher de l’équilibre entre phase vapeur et phase liquide, on utilise un dispositif de mise en contact intime appelé plateau. Une colonne de distillation est constituée par un ensemble de ces plateaux. Le liquide descendant d’un plateau au plateau suivant s’enrichit progressivement en produits lourds, c’est-à-dire, dans notre cas particulier, s’enrichit en oxygène, alors que le gaz s’élève de plateau en plateau et s’enrichit en azote. Dans le plateau de distillation, le gaz montant « barbote » dans le liquide, d’où l’existence d’une perte de charge non négligeable. Dans le garnissage, le gaz montant « lèche » la surface où le film liquide est étalé par capillarité. L’utilisation des garnissages nécessite donc:– une excellente distribution du liquide sur toute la section de la colonne;– le maintien de cette distribution lors de la descente du liquide dans la colonne (ce qui impose un garnissage organisé, et non un garnissage en vrac);– une très grande surface de contact entre gaz et liquide.Dans ces conditions, on peut obtenir une H.E.P.T. (hauteur équivalente à un plateau théorique) de 150 à 200 mm et une perte de charge quatre fois inférieure à celle des plateaux.Pour que la séparation soit complète, il faut engendrer dans la colonne un certain débit de gaz montant à partir de la base et produire un certain reflux de liquide à partir du sommet. On dit couramment qu’une colonne de distillation doit être chauffée aux pieds et refroidie à la tête, le chauffage créant la quantité de vapeur nécessaire et le refroidissement créant la quantité de liquide nécessaire pour obtenir la séparation cherchée. Dans le cas de l’air, l’originalité est due au fait que le chauffage de la cuve de la colonne s’effectue à une température de 漣 181 0C environ, cependant que le refroidissement en tête nécessite une température de 漣 194 0C.Les échangeurs de chaleurLes échangeurs de chaleur permettent de récupérer presque totalement le froid contenu dans l’oxygène et l’azote sortant de la colonne de distillation. Les échangeurs à haute performance sont des échangeurs à plaques en aluminium brasé au four sous vide, comme représenté dans la figure 11. Ils se présentent en modules de 6,5 m de longueur et de section (1,4 憐 1,4) m2. Un module peut traiter jusqu’à 30 000 nm3/h d’air.Production d’oxygèneLa pureté requise pour l’oxygène peut être, selon les cas: de 99,5 p. 100 pour la soudure, le décriquage, l’oxycoupage, les fours d’aciéries, etc.; de 95 à 99 p. 100 pour le cracking d’hydrocarbures, la gazéification du charbon, l’obtention de combustibles liquides à partir de gaz naturel, la réduction directe des minerais de fer, etc.; de 70 à 95 p. 100 pour l’enrichissement du vent des hauts-fourneaux.Dans le cas des aciéries, l’oxygène est utilisé à une pression comprise entre 12 et 15 bars, mais le débit consommé varie dans le temps, et il est nécessaire de prévoir des « poumons » fonctionnant entre 15 et 30 bars. C’est pourquoi l’oxygène est, dans ce cas, distribué à une pression de l’ordre de 30 à 40 bars.Du fait de la sûreté d’approvisionnement exigée par les consommateurs, les grandes centrales de production sont reliées entre elles par un réseau de gazoduc.Dans le cas des clients de la chimie, la consommation d’oxygène est constante, mais la pression d’utilisation est souvent plus élevée: en général de 30 à 80 bars.Production d’azoteQuand cela est possible, l’azote sera le sous-produit de la fabrication de l’oxygène. Selon les utilisations, il sera à haute pureté (1 v.p.m. d’oxygène [v.p.m. indique la teneur en volume, en partie par million]) ou sera tout simplement prélevé à la sortie de l’appareil sur l’azote résiduaire (2 p. 100 d’oxygène pour des besoins d’inertage, par exemple). Mais, très souvent, il est nécessaire de réaliser des appareils dédiés à la seule production d’azote, car trop éloignés des sites « oxygène ». On utilise alors des appareils dits H.P.N. (high purity nitrogen ) produisant directement l’azote à haute pureté et à une pression de 8 bars absolus. Les plus perfectionnés sont ceux qui sont destinés à l’industrie électronique. Les impuretés nominales admissibles en oxygène, hydrogène, oxyde de carbone, gaz carbonique, C n H m et H20 sont de 1 v.p.b. (1 partie par milliard), ce qui nécessite, en plus de la distillation, une épuration catalytique sur l’air traité. De plus, l’azote doit être pratiquement exempt de poussière, soit:– moins d’une particule de plus de 0,1 猪m par S.C.F. (standard cubic foot );– moins de 0,12 particule de plus de 1 猪m par S.C.F.;– moins de 0,05 particule de plus de 10 猪m par S.C.F.Production d’oxygène et d’azote liquideIl est également nécessaire de produire d’importantes quantités d’oxygène et d’azote liquide:– soit pour constituer des stockages de sécurité; en cas d’interruption de la production (par déclenchement électrique, par exemple), le démarrage des pompes de secours oxygène ou azote et la gazéification du liquide comprimé permettent d’assurer la continuité de l’approvisionnement en attendant le redémarrage de l’unité de production;– soit pour la distribution de l’oxygène et de l’azote liquide vers les petits clients, car c’est le seul mode de transport économique pour des distances de 30 à 300 km environ.La figure 12 représente le schéma d’une installation avec un cycle de liquéfaction intégré. La capacité de liquéfaction est généralement comprise entre 50 t/j et 1 000 t/j (soit de 1 500 à 30 000 nm3/h de gaz liquéfié). Les stockages dans les centrales peuvent aller jusqu’à 5 000 m3 en eau. Ces stockages sont à basse pression. Les stockages en clientèle sont beaucoup plus modestes, la taille nominale est de 300 m3, et la pression de tarage est en général de 15 bars absolus.Séparation des gaz autres que l’airGaz riches en hydrogèneL’hydrogène peut être utilisé sous différentes formes: combiné avec l’azote sous forme d’ammoniac, c’est une matière première fondamentale pour l’industrie chimique; à l’état pur, il peut être combiné directement dans certains processus d’hydrogénation, et sous cette forme il est consommé en grande masse dans les raffineries, où l’on voit apparaître des réseaux de canalisations d’hydrogène analogues à ceux utilisés pour l’oxygène; à l’état liquide, c’est le meilleur combustible connu pour les fusées spatiales; il peut constituer une source frigorifique intéressante pour certaines applications, par suite de son point d’ébullition intermédiaire entre celui de l’azote liquide et celui de l’hélium liquide. Par distillation à 漣 250 0C, on peut extraire le deutérium, toujours contenu, en proportion variable, dans l’hydrogène naturel. Enfin, notons que l’hydrogène est considéré par un certain nombre d’experts comme le combustible d’avenir, car il est à la fois non polluant et peut être obtenu par énergie renouvelable (électrolyse à partir d’énergie solaire).L’hydrogène est disponible dans un certain nombre de mélanges gazeux, notamment dans les gaz résiduaires de raffinerie ou de production d’éthylène, et dans le gaz des fours à coke. Il peut être produit spécialement à partir de gaz naturel, de naphta et de fuel par des procédés de reforming ou de cracking à l’oxygène.L’hydrogène est concentré par simple refroidissement entre 漣 170 et 漣 200 0C environ. En effet, tous les constituants présents se trouvent condensés en majeure partie, et le gaz restant a une teneur en hydrogène comprise entre 95 et 98 p. 100, suffisante pour la plupart des applications.Lorsqu’on désire produire de l’hydrogène à haute pureté, la meilleure solution est le P.S.A. (pressure swing adsorption ).La production d’hydrogène liquide est courante. Le stockage et le transport à l’état liquide nécessitent une isolation très poussée appelée superisolation. Elle est obtenue sous vide profond, l’interparoi étant constituée de couches successives de matériaux isolants et de matériaux réflecteurs afin de limiter les pertes par rayonnement.Traitement du gaz naturelOn a déjà signalé le développement considérable des procédés de liquéfaction du gaz naturel en vue de son transport. Mais il n’est pas exclu d’envisager aussi l’emploi du gaz naturel liquéfié comme source d’énergie pour les camions, les trains et surtout les avions supersoniques de demain. Dans ce dernier cas, en effet, le méthane liquide représente un gain de poids de près de 30 p. 100 par rapport aux combustibles classiques. En dehors de la liquéfaction, il existe de nombreux procédés de séparation du gaz naturel à basse température. À température décroissante, on note:– Le dégazolinage : par refroidissement à des températures modérées pouvant atteindre 漣 60 à 漣 70 0C, on peut condenser les hydrocarbures les plus lourds contenus dans le gaz naturel afin de les exploiter séparément. Cette récupération, généralement limitée au butane et au propane, peut aller jusqu’à l’éthane, excellente matière première pour la fabrication d’éthylène.– La désazotation : certains gaz naturels comme le gaz de Groningue, en Hollande, possèdent une teneur élevée en azote ; il peut y avoir intérêt à effectuer la séparation par distillation afin de disposer de gaz naturel débarrassé de l’azote, donc à pouvoir calorifique supérieur.– La production d’hélium : l’hélium, utilisé en grandes quantités pour la soudure et dans l’industrie spatiale et nucléaire, est présent en teneur appréciable (de 1 à 4 p. 100) dans certains gaz naturels, en particulier aux États-Unis. L’extraction d’hélium s’effectue essentiellement par refroidissement à basse température dans des installations de très grande taille. L’hélium est liquéfié couramment soit pour faciliter son transport sur de longues distances, soit pour l’utiliser directement comme source frigorifique. Les réservoirs de stockage sont « superisolés » comme dans le cas de l’hydrogène liquide.Gaz riches en éthylèneLa production d’éthylène s’est développée très rapidement depuis 1955, par suite de sa place prépondérante dans la fabrication des matières plastiques. Parmi les nombreux produits de synthèse dans la fabrication desquels l’éthylène intervient, citons: le polychlorure de vinyle; des polyesters à partir d’oxyde d’éthylène et des polyglycols; le polystyrène; le polyéthylène enfin, élaboré par les procédés à haute pression I.C.I., ou à basse pression Ziegler et Philips.Les gaz traités dans les installations proviennent de différentes sources: le gaz produit par cracking thermique d’éthane dont la teneur en éthylène est de l’ordre de 30 p. 100; le gaz produit par cracking thermique d’une coupe de naphta qui contient en moyenne 25 p. 100 d’éthylène.Le gaz obtenu par cracking est traité dans une installation à basse température en deux stades successifs: condensation de la presque totalité de l’éthylène contenu dans le gaz traité; traitement du liquide ainsi obtenu dans une série de colonnes effectuant la déméthanisation, la séparation éthylène-éthane et, éventuellement, d’autres séparations quand celles-ci sont demandées (séparation du propylène, par exemple).La capacité de production des installations de production d’éthylène atteint couramment 500 000 t/an.3. Les réfrigérateurs à dilutionLa conquête des très basses températures par les physiciens et, après eux, par les ingénieurs a été liée d’abord à la liquéfaction des gaz. Celle de l’hélium par Kamerlingh Onnes, en 1908, semblait mettre un terme à la course vers le zéro absolu (face=F0019 漣 273 0C), mais ce n’était qu’une étape. En 1933, on découvrait une nouvelle technique: la désaimantation adiabatique de certains sels paramagnétiques. Ce procédé est resté, pendant plus de trente ans, le seul moyen d’atteindre des températures inférieures à 0,2 K. On a pu ainsi refroidir des substances jusqu’au voisinage du millième de kelvin, mais ce procédé présente l’inconvénient majeur de ne permettre d’enlever qu’une quantité totale limitée de chaleur. En plus, il prohibe pratiquement les mesures sous un champ magnétique qui ramènerait le système à sa température initiale [cf. CRYOLOGIE - Cryophysique].L’isotope 3 de l’hélium (3He), obtenu en quantité notable comme sous-produit de la préparation des matériaux pour la fusion thermonucléaire, a été le point de départ d’un nouveau bond en avant. L’hélium 3 en lui-même a un point critique plus bas que l’hélium 4 (4He), et on peut, de ce fait, obtenir des températures plus basses par simple évaporation.La compression adiabatique de l’hélium 3 le faisant passer de l’état liquide à l’état solide s’accompagne d’un refroidissement en dessous de 0,3 K (cf. HÉLIUM et GAZ RARES). Cet effet ouvre la porte des millidegrés, mais, pas plus que la désaimantation adiabatique, il ne fournit de réfrigération continue.Le procédé le plus fécond est la dilution continue de l’hélium 3 dans l’hélium 4. Suggéré par H. London en 1951, défini de manière plus précise par H. London, G. R. Clarke et E. Mendoza en 1962, il fut appliqué avec succès en 1966 indépendamment par H. E. Hall et B. Neganov.Le principe du réfrigérateur à dilution consiste à enlever de la chaleur en évaporant un liquide. Le liquide est de l’hélium 3 pratiquement pur, et le rôle de la vapeur est joué par de l’hélium 3 en solution dans l’hélium 4 liquide. Avant d’étudier l’application de ce principe, il est bon de revenir sur l’origine des limites de l’évaporation classique, puis de voir ce qu’apportent les mélanges des deux isotopes de l’hélium.Limites de la réfrigération par évaporationLorsqu’on fournit de l’énergie, sous forme de chaleur, à un liquide, on élève sa température, ou encore on augmente l’énergie cinétique des atomes qui le constituent. Pour une certaine température, fonction de la pression du gaz qui surmonte le liquide, les atomes possèdent une énergie suffisante pour échapper à l’attraction des autres atomes dans le liquide; chaque atome quittant le liquide lui enlève ainsi une énergie qui est la chaleur latente de vaporisation par atome. La quantité de chaleur totale enlevée au liquide est égale à la chaleur de vaporisation multipliée par le nombre d’atomes extraits du liquide.À très basse température, le seul fluide disponible est l’hélium dont la chaleur de vaporisation varie peu lorsqu’on s’approche du zéro absolu. La seule possibilité d’augmenter la puissance de réfrigération réside dans l’augmentation de la vitesse d’extraction des atomes du liquide, c’est-à-dire du débit de la pompe aspirant les vapeurs.Malheureusement, pour une pompe donnée, le débit est proportionnel à la densité des vapeurs, densité qui tend très rapidement vers zéro lorsque la température tend vers zéro (selon une décroissance exponentielle). Ce procédé ne laisse donc aucun espoir d’atteindre de très basses températures; la limite actuelle est de 0,71 K avec l’hélium 4 et de 0,21 K avec l’hélium 3.Propriétés des mélanges d’hélium 3 dans l’hélium 4L’hélium 4 pur liquide est superfluide au-dessous d’une température de 2,17 K sous sa pression de vapeur saturante (point); si on lui ajoute des atomes d’hélium 3, la température d’apparition de la superfluidité diminue pour devenir 0,6 mK dans l’hélium 3 pur. Pour de faibles concentrations, les atomes d’hélium 3 sont séparés par un grand nombre d’atomes d’hélium 4 et les particularités spécifiques de leurs interactions ne se manifestent pas. Les atomes d’hélium 3 ont donc un comportement de gaz à l’intérieur du volume limité par la solution; ils créent en particulier une certaine pression dite pression osmotique . Ce comportement de « gaz » des atomes d’hélium 3 dans l’hélium 4 est encore rendu plus aigu par le fait qu’à très basse température l’hélium 4 superfluide n’offre aucune résistance aux déplacements des atomes d’hélium 3 et se comporte comme une sorte d’« éther ».Les isotopes 3 et 4 de l’hélium ne sont cependant miscibles en toute proportion qu’au-dessus d’une température de 0,87 K. En dessous de cette température critique de miscibilité complète, les mélanges des deux isotopes, pour certaines concentrations, se séparent spontanément en deux phases. L’une, riche en hélium 4 superfluide, se rassemble au fond du récipient dans lequel se trouve le mélange; l’autre, riche en hélium 3, plus légère, surnage. Au fur et à mesure que la température s’abaisse, la phase supérieure s’enrichit en hélium 3, la phase inférieure en hélium 4. Ce phénomène, observé pour la première fois en 1957 par V. P. Peshkov et K. N. Zinov’eva à Moscou, est une conséquence de la chaleur de mélange des deux isotopes. Près du zéro absolu de température, la phase supérieure tend vers de l’hélium 3 pur, alors que la phase inférieure existe avec une concentration limite de 6,4 p. 100 d’hélium 3. L’existence d’une solubilité finie au zéro absolu de température n’est pas en contradiction avec le troisième principe de la thermodynamique; l’application de la mécanique quantique montre que le « gaz » d’hélium 3 dans l’hélium 4 satisfait comme tous les autres corps à ce principe.Cette séparation de phase n’est pas spécifique de l’hélium et se rencontre dans d’autres mélanges (par exemple avec l’eau et le phénol qui ont une température critique de démixtion de 65 0C). Mais c’est le seul exemple connu de séparation d’isotopes.L’aspect du diagramme de phase des solutions liquides d’hélium 3 et d’hélium 4 (fig. 12 a) offre une analogie assez complète avec la courbe dite de Mathias qui donne les densités d’un gaz et d’un liquide purs à l’état saturé en fonction de la température (fig. 12 b). En particulier, les propriétés du point critique de miscibilité complète se comparent avec celles du point critique d’un corps pur.Principe du réfrigérateurÀ très basse température, le quasi-gaz d’hélium 3 est en équilibre avec l’hélium 3 à peu près pur de la phase supérieure. Un échange constant d’atomes se produit à travers la surface séparant les deux phases. Le passage d’un atome d’hélium 3 de la phase concentrée à la phase diluée est assez analogue au passage d’un atome d’une phase liquide à la phase vapeur qui le surmonte. Au cours de cette « évaporation », de la chaleur est absorbée. Cette quasi-évaporation est cependant très différente à l’évaporation d’un liquide ordinaire. D’une part, la phase « liquide » se trouve au-dessus de la phase « vapeur »; d’autre part, même au zéro absolu de température, ce quasi-gaz continue d’exister avec une densité et une pression non nulles (au zéro absolu la densité de la « vapeur » représenterait environ 8 p. 100 de la densité du « liquide » et la pression de la vapeur dépasserait 12 torrs). Il est donc possible de pomper le « gaz » et de créer du froid même très près du zéro absolu; la limitation des réfrigérateurs classiques à évaporation (fig. 13 a) n’existe pas pour la quasi-évaporation. Cependant, même en maintenant constante la vitesse d’extraction des atomes d’hélium 3 de la phase diluée, la puissance frigorifique tend vers zéro, car la chaleur de « vaporisation » ou chaleur de dissolution d’un atome d’hélium 3 dans la phase diluée tend vers zéro comme le carré de la température (elle est égale à 82 T 2 joules par mole d’hélium 3).Le fonctionnement continu exige deux opérations: d’abord l’injection d’hélium 3 à la partie supérieure de la boîte de mélange dans laquelle les deux phases sont en équilibre, ensuite l’extraction par pompage d’une quantité égale d’hélium 3 de la phase diluée (fig. 13 b). Sans autre apport de chaleur que celle du liquide entrant, la température du « gaz » sortant, donc de la boîte de mélange, ne peut être inférieure au tiers environ de la température du liquide entrant. On améliore le refroidissement en faisant précéder la boîte de mélange d’un échangeur de chaleur, dans lequel le « gaz » sortant de la phase diluée refroidit le liquide chaud entrant. Le « gaz » d’hélium 3 dans la solution diluée ne peut échapper au liquide que pour se trouver soumis aux lois communes à toutes les vapeurs, c’est-à-dire avoir une pression de vapeur tendant exponentiellement vers zéro. À cause de la très grande différence des tensions de vapeur des deux isotopes, la vaporisation de la solution diluée sépare les deux isotopes par distillation. Par exemple, à 0,6 K, une solution contenant 9 p. 1 000 d’hélium 3 (pression osmotique de 12 torrs) a une pression de vapeur de 0,035 torr, et les vapeurs sont constituées par de l’hélium 3 ne contenant que 6 p. 1 000 d’hélium 4. Pour que la distillation soit efficace, donc que la vitesse d’extraction de l’hélium 3 soit importante, elle doit s’effectuer à des températures comprises entre 0,5 et 0,7 K; ainsi la pression de vapeur n’est-elle pas trop faible. Dans ce distillateur, la concentration en hélium 4 des vapeurs ne reste faible que si le film d’hélium 4, qui grimpe le long de toute paroi en contact avec de l’hélium 4 liquide superfluide, est maintenu négligeable. De nombreux dispositifs, plus élaborés qu’un simple diaphragme, ont été mis au point pour pallier cette difficulté.À la sortie de l’échangeur, la solution diluée est envoyée dans le distillateur où l’hélium 3 est extrait de la solution. L’hélium 3 est ensuite évacué sous forme gazeuse jusqu’à l’extérieur où il est pompé et recomprimé à température ambiante à l’aide d’une pompe à vide. La solution diluée à environ 6,4 p. 100 dans la boîte de mélange est en communication avec le distillateur par l’intermédiaire de l’échangeur. L’équilibre thermodynamique des solutions veut que la pression osmotique de l’hélium 3 dans la colonne stationnaire d’hélium 4 soit sensiblement constante; cela impose à la concentration de décroître lorsqu’on chemine vers le distillateur où elle n’est plus que d’environ 1 p. 100.L’hélium 3 sortant de la pompe est réinjecté à l’intérieur du cryostat sous une pression de 30 à 50 torrs. Il est refroidi et condensé au moyen d’un bain d’hélium 4 au voisinage de 1 K. Il est refroidi plus avant par passage dans un serpentin au contact du distillateur, puis dans l’échangeur à contre-courant avec la solution diluée. Un capillaire placé au voisinage du distillateur limite le débit et assure la détente du liquide de la pression de condensation jusqu’à la basse pression régnant dans la boîte de mélange. Le liquide détendu et refroidi est enfin injecté dans la boîte de mélange. Le schéma de principe du réfrigérateur est représenté par la figure 14.La partie la plus critique du réfrigérateur est l’échangeur de chaleur entre la boîte de mélange et le distillateur. Il existe en effet, à la surface de contact entre un solide et l’hélium liquide, un saut de température lorsque de la chaleur passe de l’un à l’autre: c’est la résistance thermique de Kapitza. L’écart de température est proportionnel à la puissance transmise et inversement proportionnel au cube de la température. Il est donc nécessaire d’utiliser de très grandes surfaces d’échange tout en conservant de faibles volumes d’hélium 3 et de faibles pertes de charge. La solution actuelle se rapproche beaucoup de la solution adoptée pour les échangeurs de gros liquéfacteurs avec des plaques séparées par des feuilles ondulées (cf. supra , chap. 1, Liquéfaction et séparation des gaz ). Mais ici les épaisseurs des plaques et des canaux de circulation sont de l’ordre de la dizaine de microns.Les limites des réfrigérateurs à dilutionIl n’est malheureusement pas possible d’atteindre des températures de plus en plus basses en augmentant seulement la surface de l’échangeur de chaleur. Le quasi-gaz d’hélium 3, circulant de la boîte de mélange au distillateur à travers l’échangeur de chaleur, possède une viscosité qui croît lorsque la température décroît. Cette viscosité entraîne par frottement une chute de la pression osmotique. On assiste ainsi à une détente de Joule-Thomson qui entraîne, selon la valeur du coefficient de dilatation thermique 見 du fluide comparée à celle du gaz parfait à la même température ( 見 = -1), un refroidissement ( 見 麗 見), ou un réchauffement ( 見 礪 見). Au voisinage du zéro absolu de température, 見 devient très faible, et conduit donc toujours à un échauffement. La chaleur ainsi dégagée est transmise par conduction à la boîte de mélange. Pour réduire cet échauffement par frottement, on peut diminuer la perte de charge en augmentant la section de passage du fluide, mais alors on fait croître la conduction de chaleur à travers le liquide dans les tubes.Les limites de la température qu’on peut maintenir avec ces réfrigérateurs en fonctionnement continu dépendent donc beaucoup de l’échangeur de chaleur. Les échangeurs tubulaires simples permettent d’atteindre assez facilement quelques centièmes de kelvin. Il est possible de descendre à 2 millikelvins au moins, avec des échangeurs très élaborés comportant de la poudre d’argent frittée. Les puissances qu’on peut transmettre à ces températures de quelques millièmes de kelvin sont de l’ordre du nanowatt.Il sera sans doute possible, dans l’avenir, d’abaisser encore de quelques dix millièmes de kelvin la limite de ces appareils, mais on peut plutôt s’attendre à une augmentation des débits de fluide circulant, c’est-à-dire à une augmentation des puissances frigorifiques disponibles.
Théoriquement, un système à absorption est aussi efficace qu’un système à compression avec son moteur, pourvu que les trois températures de source se correspondent respectivement.Pour les températures supérieures à 0 0C (climatisation), on peut employer le couple eau-bromure de lithium, dans lequel l’eau est le fluide frigorigène.Si on introduit dans l’évaporateur une pression partielle p d’hydrogène de sorte que p c = p 0 + p , on élimine la pompe de circulation, et la solution circule par thermosiphon: c’est le cas des réfrigérateurs ménagers.Les systèmes à absorption sont généralement adoptés lorsqu’on ne dispose pas d’énergie mécanique, ou pour valoriser de la chaleur perdue (vapeur détendue industrielle).Évaporation et condensationÉvaporateursLes évaporateurs sont des refroidisseurs d’air ou de liquide et sont alimentés par injection directe ou regorgement.Les refroidisseurs d’air sont appelés frigorifères. L’évaporateur, compris dans un caisson, est constitué de tubes à ailettes dont l’écartement est d’autant plus grand que 0 est plus basse: de 8 à 12 millimètres en réfrigération et de 16 à 20 millimètres en congélation. L’air, aspiré par des ventilateurs en bas du local, est refroidi sur l’évaporateur et est refoulé sous le plafond (l’évaporateur des réfrigérateurs ménagers fonctionne en convection naturelle et est constitué de plaques d’aluminium).Les refroidisseurs de liquide (pour les industries alimentaires, ou pour les circuits de climatisation à eau glacée) sont constitués d’un évaporateur à tubes lisses placé dans un bac ou dans un échangeur, de conception assez semblable à celle d’un condenseur multitubulaire.Alimentation des évaporateursEn injection directe , le fluide, détendu de p c à p 0, est directement introduit dans l’évaporateur; son débit est réglé par le détendeur (détendeur thermostatique) pour qu’en sortant il soit surchauffé. S’il n’en était pas ainsi, le compresseur pourrait aspirer du liquide, et les clapets d’aspiration risqueraient de casser (coup de liquide ); en outre, s’il y a entraînement d’huile, la vitesse de la vapeur basse pression doit être d’au moins 10 mètres par seconde, pour ramener l’huile au compresseur.Ce système est simple et utilisable avec tout fluide, pour des machines de toutes puissances; cependant, en pratique, il est surtout réservé aux équipements de petite et moyenne puissance (appareils commerciaux, agricoles, petits climatiseurs). En effet, l’efficacité de l’évaporateur ne peut pas être maximale, car de la vapeur apparaît dès la détente, or les transferts sont moins bons en phase gazeuse qu’en phase liquide.Dans l’alimentation par regorgement , le fluide est détendu dans une bouteille séparatrice basse pression (B.S.B.P.); la vapeur, qui est saturée sèche, est aspirée en haut de cette bouteille, tandis que le liquide basse pression s’accumule dans le fond et est envoyé, par gravité ou pompe, dans l’évaporateur. Le fluide, sortant de ce dernier, a un excès de liquide; l’efficacité de l’évaporateur est d’autant meilleure que le titre en liquide du fluide au retour dans la B.S.B.P. est plus grand, ce qui explique l’intérêt de l’alimentation par pompe. Le détendeur, à flotteur ou électronique, règle le niveau liquide dans la B.S.B.P.L’alimentation par regorgement est réservée aux équipements industriels de grosse puissance et à ceux à régime thermique variable (tunnels de congélation, par exemple).CondenseursSelon le milieu utilisé, le condenseur est à air ou à eau. Les condenseurs à air des réfrigérateurs ménagers sont à convection naturelle; mais tous les autres appareils, de puissance commerciale ou industrielle, sont à convection forcée: l’air est aspiré par des ventilateurs à travers une batterie à ailettes serrées; le débit est important: exprimé en mètres cubes par heure, le chiffre est à peu près quatre à cinq fois plus grand que celui de la puissance frigorifique en watts.Le condenseur à eau en circuit ouvert le plus utilisé est du type multitubulaire horizontal, où l’eau fait un ou plusieurs passages dans des tubes, tandis que le fluide se condense dans l’espace compris entre l’extérieur des tubes et la virole. Comme l’échauffement de l’eau est de l’ordre de 5 à 10 0C, le débit d’eau en litres par heure est voisin du chiffre de la puissance en watts divisé par . Pour réduire la consommation, l’eau est recyclée dans un réfrigérant atmosphérique , où elle est refroidie par évaporation. La dépense est, en théorie, égale à 1 ou 2 p. 100 et, en pratique, à 5 p. 100 du débit en circuit ouvert car, pour limiter la concentration en sels, une partie de l’eau est purgée. Le condenseur et le réfrigérant atmosphérique peuvent être réunis en un seul appareil, qui est un condenseur évaporatif. Notons que, en France, les installations à condenseur à eau doivent comporter un dispositif économiseur.ApplicationsActuellement, pratiquement dans toutes les activités humaines, industrielles, agricoles, sociales, médicales, etc., interviennent des systèmes frigorifiques à fluide liquéfiable.L’application la plus ancienne concerne la conservation des denrées périssables. En 1876, le Français Charles Tellier démontrait de façon spectaculaire les possibilités du froid artificiel en transportant des carcasses de viande fraîche de Rouen à Buenos Aires.De nos jours, les techniques frigorifiques sont applicables pratiquement à toutes les denrées périssables, aux produits manufacturés altérables et tout spécialement aux plats élaborés ou cuisinés. À ce propos, il convient de souligner le succès dans tous les pays industrialisés des produits congelés et surtout surgelés. Cette dernière qualification est réservée à des produits d’excellente qualité initiale, sains, congelés rapidement jusqu’à une température de 漣 18 0C à cœur, emballés, conservés, transportés et commercialisés – en permanence à 漣 18 0C ou moins –, jusqu’à leur livraison en l’état aux consommateurs.Le respect de ces spécifications à l’échelle industrielle, clairement définies par le décret du 9 septembre 1964, est l’une des raisons du succès des produits surgelés dont la consommation en France progresse de 10 à 20 p. 100 par an depuis une vingtaine d’années. Les autres raisons majeures sont sans doute, d’une part, le prix de revient de la congélation industrielle (c’est-à-dire préalable à la distribution), qui est du même ordre de grandeur que celui de l’apertisation, et, d’autre part, les besoins énergétiques, qui sont à peu près deux fois plus petits que pour l’apertisation, et, enfin, la facilité d’emploi et la rapidité de préparation. Il est certain que la France n’a pu devenir fortement exportatrice pour toute une série de productions périssables que grâce au froid. Aussi, son équipement, réalisé depuis la Libération, est l’un des plus importants et des plus modernes du monde.Parallèlement au froid alimentaire, les techniques du conditionnement d’air se sont développées et diversifiées pour le confort de l’homme (bureaux, hôpitaux, salles de spectacles, logements, etc.) et pour la maîtrise de l’ambiance de certains locaux (d’ordinateur par exemple), par le contrôle de la température et de l’humidité relative, le filtrage d’air dans les industries textiles, papetières, de mécanique de précision, etc. Le froid est également un facteur de sécurité. Ainsi, toute centrale nucléaire comprend, pour cette raison, des machines frigorifiques puissantes.Une autre illustration remarquable des techniques frigorifiques concerne les progrès considérables accomplis en médecine et en chirurgie. Les greffes d’organes, et notamment les greffes cardiaques, impliquent son intervention. La préparation du plasma lyophilisé, qui a sauvé depuis la Seconde Guerre mondiale un grand nombre de vies humaines, comporte une phase initiale de congélation. Actuellement se développe une nouvelle chirurgie, la cryochirurgie, qui, par la congélation des tissus malades avec une sonde à basse température, permet leur ablation tout en ménageant les tissus sains avoisinant. Elle a eu de remarquables succès pour la guérison de certains cancers et tumeurs (peau, cerveau, poumons, etc.).La cryogénie a permis l’essor du trafic maritime des gaz naturels et gaz de pétrole, grâce à leur stockage à l’état liquide à la pression atmosphérique (transport par « navires méthaniers »). La cryogénie fournit également le carburant liquide nécessaire au lancement des fusées. En outre, pour l’avenir, les perspectives d’industrialisation de l’effet supraconducteur à « haute température », découvert en 1987, révèlent des possibilités d’application d’une portée naguère insoupçonnée.Enfin, par suite de la crise pétrolière, la recherche de procédés plus économes en énergie ou de récupération de chaleur, ainsi que l’exploitation de l’énergie solaire ont mis en évidence l’intérêt des pompes à chaleur, des transformateurs de chaleur et des systèmes à recompression mécanique des vapeurs qui trouvent leur origine dans la technique frigorifique. Grâce à ces procédés, le rendement de raffinage du brut pétrolier est passé de 94 à 98 p. 100.En conclusion, le froid artificiel, bien qu’il soit déjà plus que centenaire, fait encore partie aujourd’hui des techniques de pointe, en perpétuelle évolution.2. Liquéfaction et séparation des gazOn liquéfie des gaz par refroidissement à basse température soit pour obtenir des liquides directement utilisables en tant que fluide cryogénique ou parce que leur compacité facilite le stockage ou le transport, soit parce que la production du liquide constitue une étape intermédiaire permettant la séparation des composants du gaz liquéfié par condensation fractionnée ou par distillation.La liquéfaction des gaz est entrée dans sa phase industrielle au début de ce siècle grâce aux travaux de Karl von Linde et de Georges Claude. Son développement, d’abord assez lent, n’a fait que s’accélérer depuis, et une industrie très puissante et très dynamique, ayant ses techniques propres, repose sur les bases posées par ces chercheurs. On peut s’étonner que l’obtention aisée des basses températures, entre 漣 40 0C et 漣 210 0C, soit à l’origine d’une activité industrielle aussi importante. Il faut en trouver la raison dans le fait que les fameux « gaz permanents » du XIXe siècle, qui constituent les matières premières essentielles de cette industrie, sont des éléments fondamentaux puisqu’il s’agit principalement des gaz de l’air, de l’hydrogène, de l’oxyde de carbone, du méthane et du bioxyde d’azote.La séparation des mélanges gazeux peut être réalisée par différentes techniques: l’adsorption (en particulier l’adsorption à température ambiante et pression variable, dite Pressure Swing Adsorption ou P.S.A., qui s’est beaucoup développée ces dernières années), la perméation et la distillation.En fait, la technique de distillation, qui s’est développée la première, tient encore aujourd’hui une place prépondérante.Les progrès technologiques réalisés au cours des vingt dernières années ont permis de ramener les pertes thermiques, conséquence d’une distillation à basse température, à des valeurs très faibles. Ainsi, pour un appareil de séparation d’air de grande taille, la tenue frigorifique ne requiert que 6 à 7 p. 100 de l’énergie totale dépensée pour la séparation elle-même. Dans ce cas, les échangeurs de chaleur présentent des écarts minimaux de température de 1 à 3 0C, et les plateaux de distillation, des pertes de charge inférieures à 2,5 cm d’eau. Depuis 1985, une nouvelle technique de distillation par garnissage organisée s’est progressivement développée; elle permet de réduire la perte de charge par plateau théorique à moins de 1 cm d’eau.L’emploi du chalumeau oxyacétylénique, mis au point par Charles Picard en 1901, est à l’origine d’une production industrielle de l’oxygène, encore bien modeste à l’époque. La capacité des appareils de production augmenta progressivement entre les deux guerres mondiales pour atteindre quelques centaines à quelques milliers de mètres cubes par heure.L’introduction, puis la généralisation, des procédés d’élaboration de l’acier à l’oxygène a pratiquement décuplé la production d’oxygène au cours des années 1960-1970.De nouvelles utilisations de l’oxygène dans l’industrie chimique (oxyde d’éthylène, cracking d’hydrocarbures, gazéification du charbon) ont conduit à augmenter sans cesse la taille des installations.Les plus grandes unités actuelles ont été réalisées par L’Air liquide pour la gazéification du charbon dans les usines de Sasol en Afrique du Sud. La centrale d’oxygène comprend treize lignes de production de 2 250 t/j d’oxygène à une pureté de 98,5 p. 100 et à une pression de 3,5 MPa.L’important développement de réseaux de distribution par gazoduc dans les pays industrialisés, la sécurité d’approvisionnement assurée par des stockages liquides de grande capacité (de 3 000 000 à 4 000 000 l) font de l’oxygène une utilité fiable, et d’une grande souplesse d’utilisation.Parallèlement au développement industriel de l’oxygène extrait de l’air atmosphérique, l’essor simultané de l’azote obtenu dans les mêmes installations n’a pas été moins impressionnant.Les premières applications ont été la production d’ammoniac par synthèse sur catalyseur à haute pression. L’ammoniac, comme l’on sait, est constitué de trois volumes d’hydrogène pour un volume d’azote, et, dès les premières installations réalisées après la mise au point du procédé de synthèse par Haber en 1913, l’azote fut produit par distillation d’air à basse température, et l’hydrogène provenant d’un gaz à l’eau, ou d’un gaz de fours à coke, fut lui aussi purifié par refroidissement à basse température, puis « lavage à l’azote liquide ».À partir de la production d’ammoniac et de celle d’acide nitrique (R 漣H3) qui résulte de son oxydation, une vaste industrie des engrais azotés a pris naissance. Son développement rapide devait bientôt libérer l’Europe de son assujettissement aux engrais naturels étrangers, par exemple les nitrates du Chili. L’acide nitrique concentré trouvait également des débouchés dans le domaine militaire (explosifs, vésicants).Ainsi, historiquement, la liquéfaction de l’air apparaît comme l’une des contributions fondamentale à l’essor de l’industrie chimique lourde contemporaine [cf. GÉNIE CHIMIQUE].Mais l’azote, en temps que gaz neutre à bon marché, disponible si nécessaire en très grandes quantités, trouve aujourd’hui une multitude d’applications, et sa consommation ne cesse de croître.De même, l’argon, gaz rare non réactif présent dans l’atmosphère, produit simultanément à l’oxygène, voit son marché s’accroître constamment, en particulier dans le domaine de la soudure et de la sidérurgie.Outre le traitement de l’air et la purification de gaz destinés à la synthèse de l’ammoniac, les séparations cryogéniques sont nombreuses. On les rencontre par exemple:– pour purifier l’hydrogène présent dans les gaz de raffineries et dont celles-ci ont besoin pour les procédés d’hydrogénation (en particulier pour la désulfuration);– pour purifier l’hydrogène et l’oxyde de carbone dans l’industrie chimique (synthèse de l’acide acétique);– pour produire les énormes quantités d’éthylène pur nécessaires à la fabrication des matières plastiques.À côté de la séparation des gaz, leur liquéfaction joue aussi un rôle de premier plan. La diminution de volume consécutive à la liquéfaction permet en effet de stocker et de transporter facilement, à la pression atmosphérique, l’oxygène, l’azote, l’argon et l’hélium, ainsi que l’hydrogène, le méthane et l’éthylène. L’emploi de l’hydrogène liquide comme combustible des fusées spatiales a conduit les États-Unis à construire d’énormes installations de production d’hydrogène liquide. Le transport de l’hélium liquide des États-Unis vers l’Europe est devenu d’une pratique courante, malgré la température d’ébullition très basse de l’hélium (face=F0019 漣 268,9 0C), et les techniques d’isolation particulièrement coûteuses pour réduire les pertes durant le transport.L’exemple le plus spectaculaire est cependant celui de la liquéfaction du gaz naturel. Parmi les sources d’énergie, le gaz naturel est le plus difficile à exploiter par suite de l’éloignement entre pays producteurs et pays consommateurs.Lorsqu’une liaison par gazoduc est impossible du fait d’une traversée maritime, on peut envisager le transport du gaz sous forme liquéfiée à la pression atmosphérique à 漣 161 0C dans des « méthaniers ».Les usines modernes ont plusieurs lignes de liquéfaction, chacune capable de liquéfier 400 000 nm3/h de gaz. Les méthaniers ont une capacité de 135 000 m3 en eau. Les tanks fixes de stockage au départ et à l’arrivée ont une capacité du même ordre.Le trafic entre l’Alaska (Shell), Brunei, l’Indonésie, île de D s et le Japon est devenu important, de même qu’entre l’Algérie et l’Europe.Mais il existe aussi une cinquantaine d’installations d’écrêtement des pointes de consommation (peak shaving en anglais). Les gazoducs de gaz naturel, peu chargés en été, sont incapables d’assurer les pointes de consommation hivernales, dont la durée est souvent limitée à quelques jours, voire à quelques semaines. On y remédie en liquéfiant et en stockant le gaz naturel au voisinage immédiat des lieux de consommation pendant la période estivale; ensuite, on revaporise le gaz naturel ainsi stocké pendant les pointes de consommation hivernales.Production des températures de liquéfactionCycle à cascadeLe moyen de production de froid par cascade repose sur le principe du cycle frigorifique à compression de vapeur et changement de phases (cf. CRYOLOGIE - Cryogénie [production du froid]).Le cycle à cascade consiste essentiellement en une suite de cycles de réfrigération utilisant chacun le fluide frigorigène le mieux adapté afin de transférer les frigories dont on a besoin de niveau en niveau jusqu’à la température finale désirée (fig. 6).Cycle à cascade incorporéeDans le cas du cycle à cascade incorporée, les différents compresseurs du cycle à cascade sont remplacés par une machine unique qui comprime un fluide frigorigène constitué par un mélange de gaz (fig. 7). La comparaison entre le schéma de principe d’un cycle à cascade classique et celui d’un cycle à cascade incorporée met en évidence la supériorité de ce dernier, non seulement du point de vue de la simplicité, de la sécurité d’exploitation et de l’entretien mais aussi du point de vue économique.Les premières applications de ce procédé, à l’échelle industrielle, ont été réalisées à Marsa el-Brega (Libye) dès 1970 par l’Air Product, et à Skikda (Algérie) par Technip-Air liquide, où sont traités avec succès 4,5 milliards de mètres cubes de gaz naturel par an.Ce procédé est aussi dénommé procédé à « fluide frigorigène composite » ou parfois à « fluide unique ». Il a reçu de nombreuses améliorations, notamment grâce à l’apport d’une réfrigération en tête au moyen d’un cycle à propane. Il est maintenant appliqué dans la quasi-totalité des installations de liquéfaction de gaz naturel.DétenteDeux procédés de détente existent: la détente sans travail utile extérieur évoquée dans la section précédente (cf. THERMODYNAMIQUE - Thermodynamique technique), essentiellement utilisée dès l’origine dans le procédé de Karl von Linde, et la détente avec fourniture de travail par l’intermédiaire d’une machine à pistons, selon le procédé de Georges Claude. Ce procédé, plus difficile à réaliser par suite de la présence d’organes mobiles aux basses températures de l’air liquide, mais susceptible d’un meilleur rendement énergétique, s’est imposé progressivement à la suite des perfectionnements successifs de la technique (lubrification à basse température, superfinition et pistons secs, mise au point des turbines, etc.).Dans les deux cas, on favorise la liquéfaction en faisant précéder le dispositif de détente par un échangeur de chaleur à contre-courant dans lequel le gaz détendu à basse température refroidit méthodiquement le gaz chaud à haute pression.Le cycle de StirlingUn procédé de production de froid par détente avec travail extérieur fonctionnant en circuit fermé est le cycle de Stirling. Dans ce cycle, le piston du compresseur et le piston de la machine de détente sont calés à 900 sur le même arbre-manivelle, la capacité de compression et la capacité de détente étant constamment en liaison par l’intermédiaire d’un régénérateur. L’ensemble se présente donc sous une forme très compacte, sans soupape de distribution. La société Philips a développé plusieurs modèles de machines frigorifiques utilisant ce cycle d’un rendement avantageux.Séparation des constituants de l’airL’air est un mélange de corps purs constitué essentiellement des gaz indiqués dans le tableau ci-dessous.Les constituants essentiels sont visiblement l’oxygène et l’azote. Viennent ensuite quelques gaz rares à teneur constante, puis un certain nombre d’impuretés à teneur variable et dépendant en particulier de la pollution: vapeur d’eau, C2 (moyenne 300 vpM), CH4 (moyenne 5 vpM), H2 etc.DistillationLa séparation de l’air en ses constituants s’effectue par distillation. Après compression et purification, l’air traité est refroidi jusqu’à sa température de rosée, pour y être distillé (fig. 8).Les courbes représentatives sur les figures 9 et 10 donnent, pour une pression donnée:– la correspondance entre la teneur en oxygène de la phase liquide et la teneur en oxygène de la phase vapeur en équilibre;– l’évolution de la teneur en oxygène de plateau théorique en plateau théorique, dans la colonne à moyenne pression (0,6 MPa abs.) et dans la colonne à basse pression (0,14 MPa abs.).Pour se rapprocher de l’équilibre entre phase vapeur et phase liquide, on utilise un dispositif de mise en contact intime appelé plateau. Une colonne de distillation est constituée par un ensemble de ces plateaux. Le liquide descendant d’un plateau au plateau suivant s’enrichit progressivement en produits lourds, c’est-à-dire, dans notre cas particulier, s’enrichit en oxygène, alors que le gaz s’élève de plateau en plateau et s’enrichit en azote. Dans le plateau de distillation, le gaz montant « barbote » dans le liquide, d’où l’existence d’une perte de charge non négligeable. Dans le garnissage, le gaz montant « lèche » la surface où le film liquide est étalé par capillarité. L’utilisation des garnissages nécessite donc:– une excellente distribution du liquide sur toute la section de la colonne;– le maintien de cette distribution lors de la descente du liquide dans la colonne (ce qui impose un garnissage organisé, et non un garnissage en vrac);– une très grande surface de contact entre gaz et liquide.Dans ces conditions, on peut obtenir une H.E.P.T. (hauteur équivalente à un plateau théorique) de 150 à 200 mm et une perte de charge quatre fois inférieure à celle des plateaux.Pour que la séparation soit complète, il faut engendrer dans la colonne un certain débit de gaz montant à partir de la base et produire un certain reflux de liquide à partir du sommet. On dit couramment qu’une colonne de distillation doit être chauffée aux pieds et refroidie à la tête, le chauffage créant la quantité de vapeur nécessaire et le refroidissement créant la quantité de liquide nécessaire pour obtenir la séparation cherchée. Dans le cas de l’air, l’originalité est due au fait que le chauffage de la cuve de la colonne s’effectue à une température de 漣 181 0C environ, cependant que le refroidissement en tête nécessite une température de 漣 194 0C.Les échangeurs de chaleurLes échangeurs de chaleur permettent de récupérer presque totalement le froid contenu dans l’oxygène et l’azote sortant de la colonne de distillation. Les échangeurs à haute performance sont des échangeurs à plaques en aluminium brasé au four sous vide, comme représenté dans la figure 11. Ils se présentent en modules de 6,5 m de longueur et de section (1,4 憐 1,4) m2. Un module peut traiter jusqu’à 30 000 nm3/h d’air.Production d’oxygèneLa pureté requise pour l’oxygène peut être, selon les cas: de 99,5 p. 100 pour la soudure, le décriquage, l’oxycoupage, les fours d’aciéries, etc.; de 95 à 99 p. 100 pour le cracking d’hydrocarbures, la gazéification du charbon, l’obtention de combustibles liquides à partir de gaz naturel, la réduction directe des minerais de fer, etc.; de 70 à 95 p. 100 pour l’enrichissement du vent des hauts-fourneaux.Dans le cas des aciéries, l’oxygène est utilisé à une pression comprise entre 12 et 15 bars, mais le débit consommé varie dans le temps, et il est nécessaire de prévoir des « poumons » fonctionnant entre 15 et 30 bars. C’est pourquoi l’oxygène est, dans ce cas, distribué à une pression de l’ordre de 30 à 40 bars.Du fait de la sûreté d’approvisionnement exigée par les consommateurs, les grandes centrales de production sont reliées entre elles par un réseau de gazoduc.Dans le cas des clients de la chimie, la consommation d’oxygène est constante, mais la pression d’utilisation est souvent plus élevée: en général de 30 à 80 bars.Production d’azoteQuand cela est possible, l’azote sera le sous-produit de la fabrication de l’oxygène. Selon les utilisations, il sera à haute pureté (1 v.p.m. d’oxygène [v.p.m. indique la teneur en volume, en partie par million]) ou sera tout simplement prélevé à la sortie de l’appareil sur l’azote résiduaire (2 p. 100 d’oxygène pour des besoins d’inertage, par exemple). Mais, très souvent, il est nécessaire de réaliser des appareils dédiés à la seule production d’azote, car trop éloignés des sites « oxygène ». On utilise alors des appareils dits H.P.N. (high purity nitrogen ) produisant directement l’azote à haute pureté et à une pression de 8 bars absolus. Les plus perfectionnés sont ceux qui sont destinés à l’industrie électronique. Les impuretés nominales admissibles en oxygène, hydrogène, oxyde de carbone, gaz carbonique, C n H m et H20 sont de 1 v.p.b. (1 partie par milliard), ce qui nécessite, en plus de la distillation, une épuration catalytique sur l’air traité. De plus, l’azote doit être pratiquement exempt de poussière, soit:– moins d’une particule de plus de 0,1 猪m par S.C.F. (standard cubic foot );– moins de 0,12 particule de plus de 1 猪m par S.C.F.;– moins de 0,05 particule de plus de 10 猪m par S.C.F.Production d’oxygène et d’azote liquideIl est également nécessaire de produire d’importantes quantités d’oxygène et d’azote liquide:– soit pour constituer des stockages de sécurité; en cas d’interruption de la production (par déclenchement électrique, par exemple), le démarrage des pompes de secours oxygène ou azote et la gazéification du liquide comprimé permettent d’assurer la continuité de l’approvisionnement en attendant le redémarrage de l’unité de production;– soit pour la distribution de l’oxygène et de l’azote liquide vers les petits clients, car c’est le seul mode de transport économique pour des distances de 30 à 300 km environ.La figure 12 représente le schéma d’une installation avec un cycle de liquéfaction intégré. La capacité de liquéfaction est généralement comprise entre 50 t/j et 1 000 t/j (soit de 1 500 à 30 000 nm3/h de gaz liquéfié). Les stockages dans les centrales peuvent aller jusqu’à 5 000 m3 en eau. Ces stockages sont à basse pression. Les stockages en clientèle sont beaucoup plus modestes, la taille nominale est de 300 m3, et la pression de tarage est en général de 15 bars absolus.Séparation des gaz autres que l’airGaz riches en hydrogèneL’hydrogène peut être utilisé sous différentes formes: combiné avec l’azote sous forme d’ammoniac, c’est une matière première fondamentale pour l’industrie chimique; à l’état pur, il peut être combiné directement dans certains processus d’hydrogénation, et sous cette forme il est consommé en grande masse dans les raffineries, où l’on voit apparaître des réseaux de canalisations d’hydrogène analogues à ceux utilisés pour l’oxygène; à l’état liquide, c’est le meilleur combustible connu pour les fusées spatiales; il peut constituer une source frigorifique intéressante pour certaines applications, par suite de son point d’ébullition intermédiaire entre celui de l’azote liquide et celui de l’hélium liquide. Par distillation à 漣 250 0C, on peut extraire le deutérium, toujours contenu, en proportion variable, dans l’hydrogène naturel. Enfin, notons que l’hydrogène est considéré par un certain nombre d’experts comme le combustible d’avenir, car il est à la fois non polluant et peut être obtenu par énergie renouvelable (électrolyse à partir d’énergie solaire).L’hydrogène est disponible dans un certain nombre de mélanges gazeux, notamment dans les gaz résiduaires de raffinerie ou de production d’éthylène, et dans le gaz des fours à coke. Il peut être produit spécialement à partir de gaz naturel, de naphta et de fuel par des procédés de reforming ou de cracking à l’oxygène.L’hydrogène est concentré par simple refroidissement entre 漣 170 et 漣 200 0C environ. En effet, tous les constituants présents se trouvent condensés en majeure partie, et le gaz restant a une teneur en hydrogène comprise entre 95 et 98 p. 100, suffisante pour la plupart des applications.Lorsqu’on désire produire de l’hydrogène à haute pureté, la meilleure solution est le P.S.A. (pressure swing adsorption ).La production d’hydrogène liquide est courante. Le stockage et le transport à l’état liquide nécessitent une isolation très poussée appelée superisolation. Elle est obtenue sous vide profond, l’interparoi étant constituée de couches successives de matériaux isolants et de matériaux réflecteurs afin de limiter les pertes par rayonnement.Traitement du gaz naturelOn a déjà signalé le développement considérable des procédés de liquéfaction du gaz naturel en vue de son transport. Mais il n’est pas exclu d’envisager aussi l’emploi du gaz naturel liquéfié comme source d’énergie pour les camions, les trains et surtout les avions supersoniques de demain. Dans ce dernier cas, en effet, le méthane liquide représente un gain de poids de près de 30 p. 100 par rapport aux combustibles classiques. En dehors de la liquéfaction, il existe de nombreux procédés de séparation du gaz naturel à basse température. À température décroissante, on note:– Le dégazolinage : par refroidissement à des températures modérées pouvant atteindre 漣 60 à 漣 70 0C, on peut condenser les hydrocarbures les plus lourds contenus dans le gaz naturel afin de les exploiter séparément. Cette récupération, généralement limitée au butane et au propane, peut aller jusqu’à l’éthane, excellente matière première pour la fabrication d’éthylène.– La désazotation : certains gaz naturels comme le gaz de Groningue, en Hollande, possèdent une teneur élevée en azote ; il peut y avoir intérêt à effectuer la séparation par distillation afin de disposer de gaz naturel débarrassé de l’azote, donc à pouvoir calorifique supérieur.– La production d’hélium : l’hélium, utilisé en grandes quantités pour la soudure et dans l’industrie spatiale et nucléaire, est présent en teneur appréciable (de 1 à 4 p. 100) dans certains gaz naturels, en particulier aux États-Unis. L’extraction d’hélium s’effectue essentiellement par refroidissement à basse température dans des installations de très grande taille. L’hélium est liquéfié couramment soit pour faciliter son transport sur de longues distances, soit pour l’utiliser directement comme source frigorifique. Les réservoirs de stockage sont « superisolés » comme dans le cas de l’hydrogène liquide.Gaz riches en éthylèneLa production d’éthylène s’est développée très rapidement depuis 1955, par suite de sa place prépondérante dans la fabrication des matières plastiques. Parmi les nombreux produits de synthèse dans la fabrication desquels l’éthylène intervient, citons: le polychlorure de vinyle; des polyesters à partir d’oxyde d’éthylène et des polyglycols; le polystyrène; le polyéthylène enfin, élaboré par les procédés à haute pression I.C.I., ou à basse pression Ziegler et Philips.Les gaz traités dans les installations proviennent de différentes sources: le gaz produit par cracking thermique d’éthane dont la teneur en éthylène est de l’ordre de 30 p. 100; le gaz produit par cracking thermique d’une coupe de naphta qui contient en moyenne 25 p. 100 d’éthylène.Le gaz obtenu par cracking est traité dans une installation à basse température en deux stades successifs: condensation de la presque totalité de l’éthylène contenu dans le gaz traité; traitement du liquide ainsi obtenu dans une série de colonnes effectuant la déméthanisation, la séparation éthylène-éthane et, éventuellement, d’autres séparations quand celles-ci sont demandées (séparation du propylène, par exemple).La capacité de production des installations de production d’éthylène atteint couramment 500 000 t/an.3. Les réfrigérateurs à dilutionLa conquête des très basses températures par les physiciens et, après eux, par les ingénieurs a été liée d’abord à la liquéfaction des gaz. Celle de l’hélium par Kamerlingh Onnes, en 1908, semblait mettre un terme à la course vers le zéro absolu (face=F0019 漣 273 0C), mais ce n’était qu’une étape. En 1933, on découvrait une nouvelle technique: la désaimantation adiabatique de certains sels paramagnétiques. Ce procédé est resté, pendant plus de trente ans, le seul moyen d’atteindre des températures inférieures à 0,2 K. On a pu ainsi refroidir des substances jusqu’au voisinage du millième de kelvin, mais ce procédé présente l’inconvénient majeur de ne permettre d’enlever qu’une quantité totale limitée de chaleur. En plus, il prohibe pratiquement les mesures sous un champ magnétique qui ramènerait le système à sa température initiale [cf. CRYOLOGIE - Cryophysique].L’isotope 3 de l’hélium (3He), obtenu en quantité notable comme sous-produit de la préparation des matériaux pour la fusion thermonucléaire, a été le point de départ d’un nouveau bond en avant. L’hélium 3 en lui-même a un point critique plus bas que l’hélium 4 (4He), et on peut, de ce fait, obtenir des températures plus basses par simple évaporation.La compression adiabatique de l’hélium 3 le faisant passer de l’état liquide à l’état solide s’accompagne d’un refroidissement en dessous de 0,3 K (cf. HÉLIUM et GAZ RARES). Cet effet ouvre la porte des millidegrés, mais, pas plus que la désaimantation adiabatique, il ne fournit de réfrigération continue.Le procédé le plus fécond est la dilution continue de l’hélium 3 dans l’hélium 4. Suggéré par H. London en 1951, défini de manière plus précise par H. London, G. R. Clarke et E. Mendoza en 1962, il fut appliqué avec succès en 1966 indépendamment par H. E. Hall et B. Neganov.Le principe du réfrigérateur à dilution consiste à enlever de la chaleur en évaporant un liquide. Le liquide est de l’hélium 3 pratiquement pur, et le rôle de la vapeur est joué par de l’hélium 3 en solution dans l’hélium 4 liquide. Avant d’étudier l’application de ce principe, il est bon de revenir sur l’origine des limites de l’évaporation classique, puis de voir ce qu’apportent les mélanges des deux isotopes de l’hélium.Limites de la réfrigération par évaporationLorsqu’on fournit de l’énergie, sous forme de chaleur, à un liquide, on élève sa température, ou encore on augmente l’énergie cinétique des atomes qui le constituent. Pour une certaine température, fonction de la pression du gaz qui surmonte le liquide, les atomes possèdent une énergie suffisante pour échapper à l’attraction des autres atomes dans le liquide; chaque atome quittant le liquide lui enlève ainsi une énergie qui est la chaleur latente de vaporisation par atome. La quantité de chaleur totale enlevée au liquide est égale à la chaleur de vaporisation multipliée par le nombre d’atomes extraits du liquide.À très basse température, le seul fluide disponible est l’hélium dont la chaleur de vaporisation varie peu lorsqu’on s’approche du zéro absolu. La seule possibilité d’augmenter la puissance de réfrigération réside dans l’augmentation de la vitesse d’extraction des atomes du liquide, c’est-à-dire du débit de la pompe aspirant les vapeurs.Malheureusement, pour une pompe donnée, le débit est proportionnel à la densité des vapeurs, densité qui tend très rapidement vers zéro lorsque la température tend vers zéro (selon une décroissance exponentielle). Ce procédé ne laisse donc aucun espoir d’atteindre de très basses températures; la limite actuelle est de 0,71 K avec l’hélium 4 et de 0,21 K avec l’hélium 3.Propriétés des mélanges d’hélium 3 dans l’hélium 4L’hélium 4 pur liquide est superfluide au-dessous d’une température de 2,17 K sous sa pression de vapeur saturante (point); si on lui ajoute des atomes d’hélium 3, la température d’apparition de la superfluidité diminue pour devenir 0,6 mK dans l’hélium 3 pur. Pour de faibles concentrations, les atomes d’hélium 3 sont séparés par un grand nombre d’atomes d’hélium 4 et les particularités spécifiques de leurs interactions ne se manifestent pas. Les atomes d’hélium 3 ont donc un comportement de gaz à l’intérieur du volume limité par la solution; ils créent en particulier une certaine pression dite pression osmotique . Ce comportement de « gaz » des atomes d’hélium 3 dans l’hélium 4 est encore rendu plus aigu par le fait qu’à très basse température l’hélium 4 superfluide n’offre aucune résistance aux déplacements des atomes d’hélium 3 et se comporte comme une sorte d’« éther ».Les isotopes 3 et 4 de l’hélium ne sont cependant miscibles en toute proportion qu’au-dessus d’une température de 0,87 K. En dessous de cette température critique de miscibilité complète, les mélanges des deux isotopes, pour certaines concentrations, se séparent spontanément en deux phases. L’une, riche en hélium 4 superfluide, se rassemble au fond du récipient dans lequel se trouve le mélange; l’autre, riche en hélium 3, plus légère, surnage. Au fur et à mesure que la température s’abaisse, la phase supérieure s’enrichit en hélium 3, la phase inférieure en hélium 4. Ce phénomène, observé pour la première fois en 1957 par V. P. Peshkov et K. N. Zinov’eva à Moscou, est une conséquence de la chaleur de mélange des deux isotopes. Près du zéro absolu de température, la phase supérieure tend vers de l’hélium 3 pur, alors que la phase inférieure existe avec une concentration limite de 6,4 p. 100 d’hélium 3. L’existence d’une solubilité finie au zéro absolu de température n’est pas en contradiction avec le troisième principe de la thermodynamique; l’application de la mécanique quantique montre que le « gaz » d’hélium 3 dans l’hélium 4 satisfait comme tous les autres corps à ce principe.Cette séparation de phase n’est pas spécifique de l’hélium et se rencontre dans d’autres mélanges (par exemple avec l’eau et le phénol qui ont une température critique de démixtion de 65 0C). Mais c’est le seul exemple connu de séparation d’isotopes.L’aspect du diagramme de phase des solutions liquides d’hélium 3 et d’hélium 4 (fig. 12 a) offre une analogie assez complète avec la courbe dite de Mathias qui donne les densités d’un gaz et d’un liquide purs à l’état saturé en fonction de la température (fig. 12 b). En particulier, les propriétés du point critique de miscibilité complète se comparent avec celles du point critique d’un corps pur.Principe du réfrigérateurÀ très basse température, le quasi-gaz d’hélium 3 est en équilibre avec l’hélium 3 à peu près pur de la phase supérieure. Un échange constant d’atomes se produit à travers la surface séparant les deux phases. Le passage d’un atome d’hélium 3 de la phase concentrée à la phase diluée est assez analogue au passage d’un atome d’une phase liquide à la phase vapeur qui le surmonte. Au cours de cette « évaporation », de la chaleur est absorbée. Cette quasi-évaporation est cependant très différente à l’évaporation d’un liquide ordinaire. D’une part, la phase « liquide » se trouve au-dessus de la phase « vapeur »; d’autre part, même au zéro absolu de température, ce quasi-gaz continue d’exister avec une densité et une pression non nulles (au zéro absolu la densité de la « vapeur » représenterait environ 8 p. 100 de la densité du « liquide » et la pression de la vapeur dépasserait 12 torrs). Il est donc possible de pomper le « gaz » et de créer du froid même très près du zéro absolu; la limitation des réfrigérateurs classiques à évaporation (fig. 13 a) n’existe pas pour la quasi-évaporation. Cependant, même en maintenant constante la vitesse d’extraction des atomes d’hélium 3 de la phase diluée, la puissance frigorifique tend vers zéro, car la chaleur de « vaporisation » ou chaleur de dissolution d’un atome d’hélium 3 dans la phase diluée tend vers zéro comme le carré de la température (elle est égale à 82 T 2 joules par mole d’hélium 3).Le fonctionnement continu exige deux opérations: d’abord l’injection d’hélium 3 à la partie supérieure de la boîte de mélange dans laquelle les deux phases sont en équilibre, ensuite l’extraction par pompage d’une quantité égale d’hélium 3 de la phase diluée (fig. 13 b). Sans autre apport de chaleur que celle du liquide entrant, la température du « gaz » sortant, donc de la boîte de mélange, ne peut être inférieure au tiers environ de la température du liquide entrant. On améliore le refroidissement en faisant précéder la boîte de mélange d’un échangeur de chaleur, dans lequel le « gaz » sortant de la phase diluée refroidit le liquide chaud entrant. Le « gaz » d’hélium 3 dans la solution diluée ne peut échapper au liquide que pour se trouver soumis aux lois communes à toutes les vapeurs, c’est-à-dire avoir une pression de vapeur tendant exponentiellement vers zéro. À cause de la très grande différence des tensions de vapeur des deux isotopes, la vaporisation de la solution diluée sépare les deux isotopes par distillation. Par exemple, à 0,6 K, une solution contenant 9 p. 1 000 d’hélium 3 (pression osmotique de 12 torrs) a une pression de vapeur de 0,035 torr, et les vapeurs sont constituées par de l’hélium 3 ne contenant que 6 p. 1 000 d’hélium 4. Pour que la distillation soit efficace, donc que la vitesse d’extraction de l’hélium 3 soit importante, elle doit s’effectuer à des températures comprises entre 0,5 et 0,7 K; ainsi la pression de vapeur n’est-elle pas trop faible. Dans ce distillateur, la concentration en hélium 4 des vapeurs ne reste faible que si le film d’hélium 4, qui grimpe le long de toute paroi en contact avec de l’hélium 4 liquide superfluide, est maintenu négligeable. De nombreux dispositifs, plus élaborés qu’un simple diaphragme, ont été mis au point pour pallier cette difficulté.À la sortie de l’échangeur, la solution diluée est envoyée dans le distillateur où l’hélium 3 est extrait de la solution. L’hélium 3 est ensuite évacué sous forme gazeuse jusqu’à l’extérieur où il est pompé et recomprimé à température ambiante à l’aide d’une pompe à vide. La solution diluée à environ 6,4 p. 100 dans la boîte de mélange est en communication avec le distillateur par l’intermédiaire de l’échangeur. L’équilibre thermodynamique des solutions veut que la pression osmotique de l’hélium 3 dans la colonne stationnaire d’hélium 4 soit sensiblement constante; cela impose à la concentration de décroître lorsqu’on chemine vers le distillateur où elle n’est plus que d’environ 1 p. 100.L’hélium 3 sortant de la pompe est réinjecté à l’intérieur du cryostat sous une pression de 30 à 50 torrs. Il est refroidi et condensé au moyen d’un bain d’hélium 4 au voisinage de 1 K. Il est refroidi plus avant par passage dans un serpentin au contact du distillateur, puis dans l’échangeur à contre-courant avec la solution diluée. Un capillaire placé au voisinage du distillateur limite le débit et assure la détente du liquide de la pression de condensation jusqu’à la basse pression régnant dans la boîte de mélange. Le liquide détendu et refroidi est enfin injecté dans la boîte de mélange. Le schéma de principe du réfrigérateur est représenté par la figure 14.La partie la plus critique du réfrigérateur est l’échangeur de chaleur entre la boîte de mélange et le distillateur. Il existe en effet, à la surface de contact entre un solide et l’hélium liquide, un saut de température lorsque de la chaleur passe de l’un à l’autre: c’est la résistance thermique de Kapitza. L’écart de température est proportionnel à la puissance transmise et inversement proportionnel au cube de la température. Il est donc nécessaire d’utiliser de très grandes surfaces d’échange tout en conservant de faibles volumes d’hélium 3 et de faibles pertes de charge. La solution actuelle se rapproche beaucoup de la solution adoptée pour les échangeurs de gros liquéfacteurs avec des plaques séparées par des feuilles ondulées (cf. supra , chap. 1, Liquéfaction et séparation des gaz ). Mais ici les épaisseurs des plaques et des canaux de circulation sont de l’ordre de la dizaine de microns.Les limites des réfrigérateurs à dilutionIl n’est malheureusement pas possible d’atteindre des températures de plus en plus basses en augmentant seulement la surface de l’échangeur de chaleur. Le quasi-gaz d’hélium 3, circulant de la boîte de mélange au distillateur à travers l’échangeur de chaleur, possède une viscosité qui croît lorsque la température décroît. Cette viscosité entraîne par frottement une chute de la pression osmotique. On assiste ainsi à une détente de Joule-Thomson qui entraîne, selon la valeur du coefficient de dilatation thermique 見 du fluide comparée à celle du gaz parfait à la même température ( 見 = -1), un refroidissement ( 見 麗 見), ou un réchauffement ( 見 礪 見). Au voisinage du zéro absolu de température, 見 devient très faible, et conduit donc toujours à un échauffement. La chaleur ainsi dégagée est transmise par conduction à la boîte de mélange. Pour réduire cet échauffement par frottement, on peut diminuer la perte de charge en augmentant la section de passage du fluide, mais alors on fait croître la conduction de chaleur à travers le liquide dans les tubes.Les limites de la température qu’on peut maintenir avec ces réfrigérateurs en fonctionnement continu dépendent donc beaucoup de l’échangeur de chaleur. Les échangeurs tubulaires simples permettent d’atteindre assez facilement quelques centièmes de kelvin. Il est possible de descendre à 2 millikelvins au moins, avec des échangeurs très élaborés comportant de la poudre d’argent frittée. Les puissances qu’on peut transmettre à ces températures de quelques millièmes de kelvin sont de l’ordre du nanowatt.Il sera sans doute possible, dans l’avenir, d’abaisser encore de quelques dix millièmes de kelvin la limite de ces appareils, mais on peut plutôt s’attendre à une augmentation des débits de fluide circulant, c’est-à-dire à une augmentation des puissances frigorifiques disponibles.
Encyclopédie Universelle. 2012.